RESEARCH ARTICLE
Physiologic Activity of Bisphosphonates – Recent Advances
Ewa Chmielewska*, Paweł Kafarski
Article Information

Identifiers and Pagination:
Year: 2016Volume: 3
First Page: 56
Last Page: 78
Publisher Id: PHARMSCI-3-56
DOI: 10.2174/1874844901603010056
Article History:
Received Date: 14/9/2015Revision Received Date: 12/4/2016
Acceptance Date: 25/04/2016
Electronic publication date: 30/05/2016
Collection year: 2016
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution-Non-Commercial 4.0 International Public License (CC BY-NC 4.0) (https://creativecommons.org/licenses/by-nc/4.0/legalcode), which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
Abstract
Background:
Bisphosphonates are drugs commonly used for the medication and prevention of diseases caused by decreased mineral density. Despite such important medicinal use, they display a variety of physiologic activities, which make them promising anti-cancer, anti-protozoal, antibacterial and antiviral agents.
Objective:
To review physiological activity of bisphosphonates with special emphasis on their ongoing and potential applications in medicine and agriculture.
Method:
Critical review of recent literature data.
Results:
Comprehensive review of activities revealed by bisphosphonates.
Conclusion:
although bisphosphonates are mostly recognized by their profound effects on bone physiology their medicinal potential has not been fully evaluated yet. Literature data considering enzyme inhibition suggest possibilities of far more wide application of these compounds. These applications are, however, limited by their low bioavailability and therefore intensive search for new chemical entities overcoming this shortage are carried out.
INTRODUCTION
Bisphosphonates were first synthesized in the 19th century, and the history of their discovery was not without some drama [1]. Soon, these compounds had been found to act as strong metal ion complexones [2] with useful industrial and household applications, including detergents, water treatment agents, dispersants preventing re-disposition of insoluble inorganic matter and compounds avoiding the metal-catalyzed decomposition of hydrogen peroxide in bleaching formulations [3].
In the early 1960s, inorganic pyrophosphate was found to act as a natural inhibitor of calcification by its interaction with hydroxyapatite. This finding made it interesting for pharmacologic applications in the treatment of medical states related to bone resorption. Unfortunately, pyrophosphate is metabolically unstable because it is rapidly hydrolyzed in the gastrointestinal tract. Thus, seeking more stable compounds, attention turned to its analogs, bisphosphonates. Similarly to pyrophosphate, bisphosphonates exhibit high affinity for bone hydroxyapatite and effectively prevent calcification. Today, these compounds have become a powerful family of pharmaceuticals for the treatment of skeletal complications of malignancy, Paget’s disease, osteoporosis, multiple myeloma, hypercalcemia and fibrous dysplasia. Their applications, their clinical implications and their mechanism of action have been reviewed in detail [4-14].
Therefore, in this review, recent studies on the biological activity of bisphosphonates will be presented with some general background provided if necessary.
ANTI-RESORPTIVE BISPHOSPHONATES
It is estimated that osteoporosis affects 200 million women worldwide and causes a huge personal and economic problems. In Europe, disability caused by osteoporosis surpasses that caused by cancer (with the exception of lung cancer) and is proportional to or even exceeds that lost to a variety of chronic non-communicable diseases, such as rheumatoid arthritis, asthma and high blood pressure-related heart disease [15].
The treatment of osteoporosis consists of lifestyle measures and pharmacologic therapy. Approval by Food and Drug Administration (FDA) of alendronate in 1995 resulted in widespread use of bisphosphonates in clinical practice. Today, numerous members of this class of compounds are available as drugs. It is evident that over 50 years, the bisphosphonates revolutionized the treatment of osteoporosis being first-line therapy for the postmenopausal-type of this disease.
Early-generation bisphosphonates differ from later-generation bisphosphonates (representative examples are shown in Scheme (1) by the absence of a nitrogen atom in their structures. It was found further that nitrogen-containing bisphosphonates are able to inhibit bone resorption 100 to 10,000 times more effectively than non-nitrogen ones, causing nearly complete displacement of the latter compounds from therapy [16].
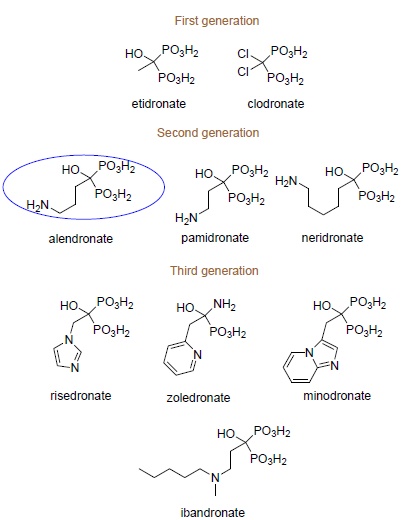 |
Scheme (1). Clinically used bisphosphonates. |
Drug efficacy is governed by its potency to inhibit farnesyl pyrophosphate synthase (FPPS), a primary target for the bisphosphonates) and its affinity to bone mineral. The latter influences uptake and retention of the drug by the skeleton, differential distribution with the bone and diffusion through the osteocyte lacunar-canalicular system [17].
Over the years, a huge variety of bisphosphonates have been obtained, and their influence on the process of bone resorption was evaluated. Because of the efficiency of currently used drugs, not much research is now being done on the design, synthesis and evaluation of new drug candidates. The large amount of data considers rather modes of their applications, longevity of treatment, recommendations and side-effects [18-20]. Most important is the evaluation of the risk of developing bisphosphonate-related osteonecrosis of the jaw – a fatal side effect of chronic use of medication [21].
Two types of efforts have been undertaken in the last decade to design new bisphosphonate anti-osteoporotic agents. The first effort is based on knowledge of the three-dimensional structure of human farnesyl pyrophosphate synthase (hFPPS) [22-24]. FPPS controls intracellular levels of farnesyl pyrophosphate, and thus, the process of protein farnesylation, which is critical for the proper subcellular localization and function of many proteins, including small GTPases that regulate a wide variety of cellular processes. In the case of bone growth modulation, inhibitors of this enzyme block excessive bone resorption in osteoclasts by causing apoptosis. The availability of the crystal structures of structurally variable bisphosphonates bound to the human enzyme allowed for the determination of its structural requirements [5, 25-27]. The knowledge of three-dimensional requirements of the enzyme active and binding sites, in turn resulted in the rationale (mostly computer-aided) design of novel effective inhibitors (representative structures are shown in Scheme 2) [5, 27-35]. However, none of these inhibitors were competitive with drugs already used to treat osteoporosis because most research effort was concentrated on finding possible anti-cancer rather than bone anti-resorptive agents.
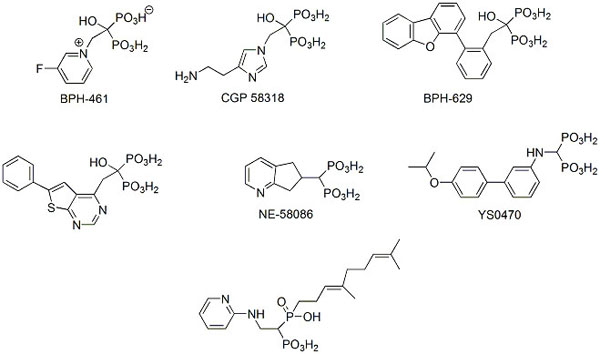 |
Scheme (2). Representative examples of bisphosphonates designed as human FPPS inhibitors. |
Because some of the side-effects caused by bisphosphonates (especially jaw osteonecrosis) may be a result of their permanent binding to bone tissue, there has been interest in the development of more non-polar compounds and even non-bisphosphonate FPPS inhibitors [36, 37]. Such inhibitors should have also higher bioavailability and thus, are also considered better candidates for anti-cancer agents. The most successful implementation of this approach was the discovery of phosphonocarboxylate cognates of risendronate and minodronate (Scheme 3). These compounds inhibit bone resorption in vivo, however, to a lesser extent than the parent drugs [5, 32, 38-40]. Later, it was found that they act as inhibitors of Rab geranylgeranyl transferase and prevent geranylgeranylation of small Rab GTPases [5, 40, 41]. Thus, these compounds might be considered as a novel class of anti-cancer agents.
Since the hydroxyl group present in most commercialized drugs enhances their binding to bone, its removal from the molecule may result in weaker binding and shorter persistence in bones. Incadronate, also called cimadronate (Scheme 3), is a drug in development for treatment of osteoporosis and hypercalcemia and is a successful example of such reasoning [42]. This approach has also been applied to design strong inhibitors of FPPS from various sources,[5, 25, 43-46] with some of them showing unusual patterns of enzyme binding [47]. Interestingly, albeit not fully developed, the approach of replacing the hydroxyl group by fluorine atom has been tested [48]. Representative structures of these two classes of compounds are also shown in Scheme (3).
 |
Scheme (3). Bisphosphonates with reduced hydrophilicity. |
Another approach was to build up libraries of amino-methylenebisphosphonates and screen their ability to inhibit proliferation of macrophage-like J774E cells [49-52]. This choice of a screening system seems to be reasonable because this cell line originates from the same precursors as osteoclasts. Although some of the studied compounds inhibited proliferation of the cells quite potently (representative structures are shown in Scheme 3), preclinical studies performed on sheep indicated that there is no direct relationship between the results of screening and the efficiency of medication in animals with induced osteoporosis [52].
An increase in hydrophobicity of bisphosphonates may also be achieved by modulation of the organic part of the molecule. Examples of a successful implementation of this idea include introduction of long hydrocarbon or aromatic substituents into molecules with previously reported activity (Scheme 4) [5, 25, 30, 31, 53, 54]. Interestingly, hydroxy-bisphosphonic analogs of bile acids appeared to be exceptionally active against L929 cells and cultures of osteoclasts. The lipophilic compounds act similarly as other bisphosphonates and additionally are more bioavailable [55]. Therefore, their influence on prenylation is not limited to bone cells and might be considered as drugs affecting multiple processes, or multi-target drugs.
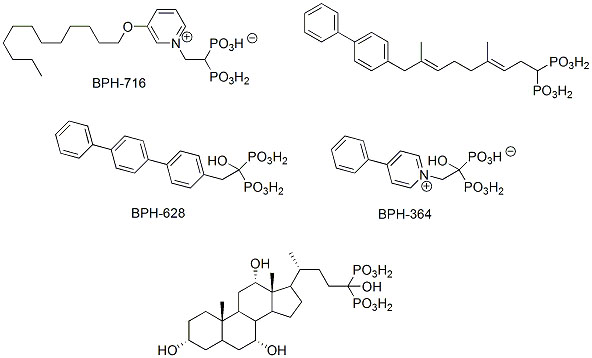 |
Scheme (4). Lipophilic bisphosphonates. |
An obvious solution to reduce bone affinity and increase bisphosphonate bioavailability is its esterification, which would mask the negative charge of phosphonic groups [56-60]. The resulting esters should also act as pro-drugs being hydrolyzed within body fluids. Although some of the esters exerted anticancer activity in cell cultures, this effort did not bring promising results so far.
An important factor influencing the anti-resorptive action of bisphosphonates is their affinity toward bones. Although intensively studied using various techniques and theoretical approaches, [61-66] this process is not fully understood. Binding is mainly mediated by the presence of two phosphonic groups, but other structural features are also important. This is reflected by a high dependence of bone affinity on the chemical structure of these drugs.
The efficiency of bisphosphonates as anti-resorptive drugs is governed by their molecular mechanism of action, affinity to bones and distribution within this tissue. The latter was studied using either radioactive or fluorescent labeled drugs [2, 67-71]. These studies indicated that distribution is also dependent on the chemical features of the drugs.
In summary, better understanding of the molecular mode of action, an exceptional selectivity of bisphosphonates for bone mineral and the process of their distribution would explain their clinical features and creates new opportunities for further discoveries.
Another, albeit poorly developed, idea is the possible use of bisphosphonates for osseointegration of implants by coating their surface with these drugs and for healing of segmental bone fractures [72-76]. The latter system requires development of a local delivery system that is easy to handle by surgeons (most likely application via syringe) in a form that would remain in the targeted area of the affected tissue. Specific glues composed of hydroxyapatite and calcium phosphate microspheres or obtained by encapsulation of bisphosphonates in hydrogels have been designed for this application [77-83].
It should be noted, however, that bisphosphonates enhance bacterial adhesion and biofilm formation on the surface of bone hydroxyapatite. This may limit their use as osseointegrating agents [84, 85].
BISPHOSPHONATES AS BONE-TARGETING UNITS
The standard routes of administration for bisphosphonates used in clinical practice are either oral or intravenous infusion. Oral administration of bisphosphonates is complicated by poor bioavailability (generally below 1%) and poor gastrointestinal tolerability [86, 87]. Due to their avid affinity to bone, between 30% and 60% of the absorbed substance rapidly binds to bone mineral. This feature was used to construct drug-bisphosphonate conjugates, which might be considered as a promising method for selective drug targeting to the bone [88, 89]. Such an active transport of therapeutic agents to bone is called osteotropic drug delivery system (ODDS). It reduces drug toxicity and improves its bioavailability at the desired site. Bisphosphonates also have an advantage over other molecules because their affinity to bone may be tuned by variation of their chemical structures.
This system was also used directly for construction of bone-regenerative drugs. Parathyroid hormone (PTH) is an 84-amino acid polypeptide that plays an important role in calcium regulation and bone remodeling. Its 34-amino acid analog Teriparatide [90] retains most of the functions of PTH and is an FDA approved drug against osteoporosis. However, being a peptide, it is unstable in body fluids and is readily hydrolyzed by proteinases. Conjugation of this compound to hydrazine bisphosphonates of varying lengths and hydrophobicity was proposed as a way to improve the therapeutic properties of Teriparatide [91]. Another approach was to conjugate a popular anti-osteoporetic drug, pamidronate, with an edible polysaccharide, pullulan, and use this system for targeting fluorescent and magnetic resonance probes. In this manner, a theranostic system was obtained [92].
One of the major reasons of cancer-related women's death is the development of bone metastases. Therefore, the selective targeting of anti-cancer drugs to bone tissue should improve their pharmaceutic performance. For example, polymers conjugated with both bisphosphonates and anti-cancer paclitaxel have been designed [93-97] to synergistically combine the anti-mitotic effect of paclitaxel with the anti-angiogenic and bone-targeting properties of bisphosphonates [98]. Furthermore, direct conjunction of bisphosphonates with popularly used anti-cancer drugs such as camptothecin, [99] bortezomib, [100] doxorubicin [101] or gemcitabine [102] has been used to target these drugs to bone tissue or to multiple myeloma cells. Representative chemical structures of such conjugates are shown in (Scheme 5).
 |
Scheme (5). Examples of conjugates of bisphosphonates with anti-cancer drugs. |
Another solution is to use conventional drug delivery systems, such as liposomes [103-106] or polymeric (preferably biodegradable) nanoparticles [107-110] with surfaces functionalized (incrusted) with bisphosphonates. Construction of such systems is challenging and thus, data on their functional activity are scarce.
Breast microcalcifications are found in about half of all women over the age of 50. They are a natural result of breast aging and are not usually due to cancer but can be a sign of pre-cancerous changes or early breast cancer if a group is found in one area and therefore, are diagnosed by mammography. Diagnosis, however, is limited to the sensitivity and specificity of this technique. Therefore, bisphosphonate-functionalized gold nanoparticles have been designed and used for contrast-enhanced radiographic studies of microcalcifications [111]. Similar approaches using specially designed technetium-99m and rhenium-188 complexes with pedant bisphosphonate groups have been applied for imaging of arterial calcification, [112] a symptom of cardiovascular disease.
Osteomyelitis is a serious bone infection most often caused by bacteria. The most common treatments are antibiotics and surgery to remove portions of bone that are infected or dead. Thus, selective targeting of antibiotics to bone seems to be profitable, and some preliminary attempts to design and obtain osteotropic systems with fluoroquinones, [113, 114] vancomycin [115] and rifamycin [116] as active components have been undertaken.
An interesting system for the treatment of kidney stones has been proposed recently. Kidney stones are endemic, and the use of extracorporeal shock wave lithotripsy where focused shock waves were used to fragment these stones have been studied [117]. The shockwaves induce the formation of cavitation bubbles, whose collapse releases energy at the stone, resulting in fragmentation into pieces small enough to be passed spontaneously. Because there is substantial amount of hydroxyapatite in these stones, bisphosphonates are good candidates to deliver microbubbles into or near urinary stones. Thus, a system has been designed where perfluoropropane is encapsulated in lipid microspheres and delivered to the kidney by a catheter. The lipidic surface is composed of the mixture of commercial lipid (DPCC) and lipid functionalized with bisphosphonate (Scheme 6). This mixture binds to the surface of the stone, and application of external ultrasonic irradiation causes stone fragmentation.
 |
Scheme (6). Bisphosphonate-based system for the delivery of gas microbubbles into kidney stones. |
ANTI-CANCER ACTIVITY
As estimated, farnesylation or geranylgeranylation account for more than 2% of the human proteome. Farnesylation of the small GTP-binding proteins, Ras, is indispensable to regulate proliferation, invasive properties and pro-angiogenic activity in human cancers [118]. Nitrogen-containing bisphosphonates are inhibitors of human FPPS and related enzymes of the mevalonate pathway and also inhibit Ras farnesylation. Thus, they should be considered as possible anti-cancer agents. This reasoning is supported by the ability of these compounds to suppress proliferation of cancer cells of prostate, [119] breast, [120, 121] melanoma, [122] ovarian [123] and colorectal cancers, [124] as well as glioblastoma [125] and multiple myelanoma [126]. Additionally these drugs have also been shown to kill cancers in humans independently on their action on bones.
The most developed strategy is the use of bisphosphonates as adjuvants in breast cancer therapy. These compounds are the major weapon to prevent bone-loss and skeleton-related events (bone pain, pathologic fractures, spinal cord compression and hyperglycemia), which accompany breast cancer therapy with aromatase inhibitors [127-129].
Interestingly, epidemiological studies seem to reveal beneficial and preventive effects of anti-osteoporetic therapy on cancer development [130, 131]. Nonetheless, the mechanisms underscoring these anti-cancer actions are not well understood. This is because the physiologic mechanism of bisphosphonate action is complex, and they have been shown to block tumor growth independently of FPPS inhibition, namely through γ,δ T-cell receptor activation, [132, 133] NF-κB inhibition, [134]. VEGF and hypoxia inducible factor-α suppression [135] as well as inactivation of epidermal growth factor receptors [136]. There is also a report showing that bisphosphonates target the three most epigenetic cell levels, namely DNA methylation, histone deacetylation and microRNAs [137].
Recent study have shown that the use of carrier technology may convert antiresorptive zoledronate into manticancer therapeutic [210]. Reformulation of calcium zolendronate into nanoscale metal-organic framework, functionalized with folate (as a selective carrier to cancer cells) served for this purpose.
Nitrogen-containing bisphosphonates are limited as anti-cancer agents because of high bone affinity and low plasma concentration. Therefore, it will be necessary to use them in combination with other drugs or search for new chemical entities.
The strategy to increase compound plasma level relies on the synthesis of new agents with increased hydrophobicity. This approach has already been described.
Another approach is to search for completely new mechanisms of action. This is offered by the use of bisphosphonate complexes with polyoxometalates [138]. These complexes can act as dual inhibitors, as phosphonate inhibits FPPS, whereas polyoxometalate interferes with redox reactions. However, the mechanism of action of some of these complexes is independent of mevalonic pathway inhibition, which suggests action via an unknown mechanism [139-141].
There are limited reports on the use of bisphosphonates as agents that are synergistic with other drugs. A recent report on the use of lovastatin, zoledronate and digeranylbisphosphonate (Scheme 7) indicated that the simultaneous use of these three compounds as inhibitors of three subsequent steps in geranylgeranyl synthesis led to the inhibition of growth and induction of apoptosis in human chronic myelogenous leukemia cells [142, 143].
 |
Scheme (7). Three-component drug against myelogenous leukemia. |
Being the most common source of cancer-related pain, bone metastases reduce functional capacity of patients and undermine their quality of life. Pain may be a result of disruption of tissue upon invasion and/or the pressure of tumorous tissue on nerve endings. Bone metastases can also activate pain receptors in pathologically changed bones. In the cases when pain is not manageable with analgesics it is commonly medicated by radiation therapy.
The efficiency of bisphosphonates in reducing pain from bone metastases is well-documented [144-146]. Zoledronic acid applied intravenously alone or complexed with radioactive metal ions has demonstrated the broadest clinical activity [147-149].
Complex Regional Pain syndrome type is a disease for which no gold-standard treatment exists to date. It may initially affect an arm or leg and spread throughout the body. Bisphosphonates offer some hope here, although the studies on their application are in infancy [150, 151].
ANTI-PROTOZOAL BISPHOSPHONATES
Protozoan infections are so called the “world's neglected diseases,” despite the fact that they affect millions of humans, spreading mostly in the poorest populations. The most important infections are leishmaniases, affecting 12 million inhabitants and Chagas disease with 8 million people affected worldwide [152]. Furthermore, malaria, which is a serious life-threatening infection caused by five different species of protozoa of the genus Plasmodium, is a huge public health concern [153]. According to the WHO report, there were 200 million malaria cases in 2013, with over half a million deaths.
Protozoan farnesyl pyrophosphate synthase (FPPS) is also a valuable target in the search for drugs against protozoa infections, and consequently, bisphosphonates were quite intensively studied here. The initial efforts had been concentrated on screening the influence of various series of these compounds against groups of parasites, such as Leishmania, [154, 155] Plasmodium, [46, 154] Trypanosoma, [154, 156, 157] Toxoplasma, [54, 154, 158] Schistosoma [159] and Cryptosporidium [160]. Thus, intensive screening of large libraries of structurally diverse bisphosphonates resulted in selection of the most effective structures and determination of the molecular mechanisms of their activities. Special emphasis have been put on bisphosphonate interactions with protozoan FPPSs and the related enzyme, namely geranylgeranyl phosphate synthase (GGPPS) [45, 161-167]. These studies have also been concentrated on the synthesis of more effective inhibitors, specifically those of increased lipophilicity appeared to be effective (Scheme 8).
 |
Scheme (8). Representative anti-protozoal bisphosphonates. |
Bisphosphonates also appeared to inhibit other protozoan enzymes, namely hexokinase [168] and squalene synthetase [169]. Additionally, their interesting action on protozoan acidosomes, acidic organelles rich in calcium and phosphorus, have been observed [170]. These findings suggest that bisphosphonates might be considered as multi-target drugs.
The soil-dwelling amoeba Dictyostelium discoideum is known for its remarkable life cycle, which makes it an ideal model organism to study a range of biological problems, such as the mechanisms of action of both first generation [171, 172] and nitrogen-containing bisphosphonates [173]. These studies indicate that bisphosphonates are able to cross peroxisomal membranes before they can exert inhibitory action.
These studies also indicated that this class of compounds might be useful as anti-amoebic agents. This was supported by their inhibitory action towards Entamoeba histolytica, the causative agent of amebiasis and Naegleria fowleria, a vector of primary amebic meningoencephalitis [174, 175]. This was an important finding because the annual number of E. histolytica infections throughout the world is estimated to be approximately 50 million.
ANTIBACTERIAL BISPHOSPHONATES
Antibiotics have always been considered one of the greatest discoveries of the 20th century. This is true, but the real interest is in the rise of antibiotic resistance [176]. In the last two decades, multi-drug resistant microorganisms challenged the scientific community into developing new antimicrobial compounds. Isoprenoid biosynthesis is an important target here because isoprenoids are involved in the early steps of bacterial cell-wall biosynthesis. New compounds affecting cell wall biosynthesis are very promising because they could also cause restoration of sensitivity to existing drugs. Therefore, it is not surprising that a huge library of bisphosphonates were tested as inhibitors of Escherichia coli growth, with some of them exhibiting quite promising activities [177-180]. Additionally, they appeared to be synergistic with other phosphonate antibiotics, such as fosfomycin.
Some of the bisphosphonates designed as FPPS inhibitors appeared to also inhibit undecaprenyl diphosphate synthase, [181, 182] an enzyme that catalyzes the sequential condensations of a farnesyl pyrophosphate with eight isopentenyl pyro-phosphates, which results in the formation of new cis-double bonds and gives undecaprenyl pyrophosphate, a metabolite serving as a lipid carrier for peptidoglycan formation in the bacterial cell wall. Systematic molecular modeling based on crystallographic studies enabled the definition of structural requirements for its inhibitors [183].
Finding that two structurally similar bisphosphonates (Scheme 9) are able to inhibit different Mycobacterium tuberculosis enzymes, decaprenyl diphosphate synthase and tuberculosinol synthase, suggest that bisphosphonates could act as multi-targeting agents [184]. This possibility was confirmed by the ability of one of the inhibitors of undecaprenyl pyrophosphate synthase, shown in Scheme (9), to additionally intercalate with DNA [185].
Other enzymes have also been considered as targets for specific antibacterial agents, namely thymidyltransferase [186] and δ1-pyrroline-5-carboxylate reductase as an inhibitor of Streptococcus pyogenes , [187] which is the cause of many important human diseases ranging from mild superficial skin infections to life-threatening systemic infections. Additionally, glutamine synthetase could also be a target of a selective anti-tuberculosis agent for use against infections of the musculoskeletal system [188].
Preparation of hydroxyphosphonate derivatives of ciprofloxacin and moxifloxacin (representative structure is shown in Scheme 9) represents a similar concept of combining bone-targeting and antibacterial properties in one molecule [189].
 |
Scheme (9). Chosen antibacterials. |
In summary, the application of bisphosphonates as potential antibacterial agents has been considered very recently, but the number of reports is limited. The design and synthesis of specific chemicals inhibiting target enzymes of chosen microorganisms is a solution.
ANTIVIRAL ACTIVITY
The finding that keto- and diketo-acids bind to the Mg(II)/Asp domain of HIV integrase in a manner similar to that observed upon their binding with prenyl transferases stimulated interest in possible use of bisphosphonates as its inhibitors [178]. This resulted in the synthesis and evaluation of several inhibitors of this enzyme (Scheme 10) [190-192]. However, integrase is a difficult target for the development of efficient anti-HIV drugs. This results from the fact that the compounds of interest have to be transported to the nuclei of infected cells, and polar bisphosphonates cannot be successfully transported.
There are also selected reports on the action of these compounds on HIV reverse transcriptase (Scheme 10)[191, 193].
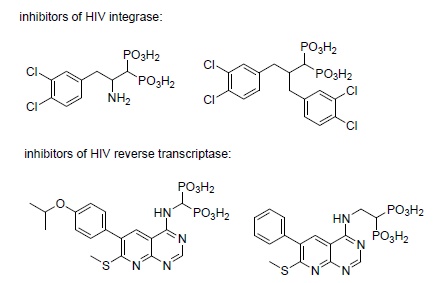 |
Scheme (10). Chosen antibacterials. |
In considering the above information, it is not surprising that pamidronate was shown to inhibit influenza virus infections in mice [194].
PLANT GROWTH REGULATORS
The herbicidal effects of bisphosphonates were first discovered in 1979 according to the patent literature, but this did not attract much attention until 1995 [195]. Their herbicidal action was then intensively studied, and they were the first reported inhibitors of farnesyl pyrophosphate synthetase (FPPS) [196, 197]. This inhibitory activity was observed when studying bleaching herbicides influencing biosynthesis of caretonoids. Further studies have shown that herbicidal aminomethylene-bisphosphonates may be considered multi-target substances, which was documented by their inhibitory activities towards glutamine synthetase, [198, 199] 3-deoxy-D-arabinoheptulosonate-7-phosphate (DAHP) synthase, [200] δ1-pyrroline-5-carboxylate (P5CR) reductase [201, 202] and pyrophosphatase [203]. Thus, these compounds may be considered a heterogeneous group of compounds with variable modes of action (Scheme 11). Quite interestingly, most active compounds contain a halogen atom or atoms in the N-aromatic substituent (Scheme 11).
Unfortunately, despite their excellent action under laboratory conditions, they have not been introduced to agriculture. Thus, recent and scarce studies on the influence of these compounds on plant growth used bisphosphonates as tools to study the role of isoprenoid biosynthesis [204-207]. These compounds enabled us to determine the high elasticity of the chloroplastic isoprenoid pathway.
 |
Scheme (11). Plant growth regulating bisphosphonates. |
Alpine pennycress (Noccaea caerulescens)is native to the mountains of central and southern Europe. It arrived in Finland at the end of the 19th century and is now used as a hyperaccumulator of zinc, cadmium and nickel ions for phytoremediation purposes [208]. The simultaneous use of the water insoluble, extremely effective metal chelator, hydroxyundecylidene-1,1,-bisphosphonic acid (Scheme 11), and this plant caused a significant increase of the metal sequestering properties of Alpine pennycress [209].
CONCLUSION
Bisphosphonates are mostly recognized by their profound effects on bone physiology. They inhibit bone resorption by inducing apoptosis of osteoclasts and thus, preventing age-related bone loss and deterioration of bone microarchitecture. Because some advanced cancers, such as breast or prostate cancer, can spread to the bone, bisphosphonates could modify the process of metastasis. Furthermore, bisphosphonates possess other useful physiologic properties, which make them promising anti-cancer, anti-protozoal, antibacterial and antiviral agents. However, their high polarity and thus low absorption in humans limit these applications. Therefore, the careful design of their chemical structures devoted to specific applications is required.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
The work was financed by a statutory activity subsidy from the Polish Ministry of Science and Education for the Faculty of Chemistry of Wrocław University of Technology. Linguistic correction was financed by Wrocław Center of Biotechnology, program The Leading National Research Center (KNOW) for years 2014-2018.