RESEARCH ARTICLE
Formulation and Pharmacokinetics of Flurbiprofen Sublimated Fast Dissolving Tablets#
Sateesh K. Vemula1, 2, *, Santhosh G. Reddy3
Article Information
Identifiers and Pagination:
Year: 2015Volume: 2
First Page: 56
Last Page: 65
Publisher Id: PHARMSCI-2-56
DOI: 10.2174/1874844901502010056
Article History:
Received Date: 21/3/2015Revision Received Date: 1/8/2015
Acceptance Date: 12/10/2015
Electronic publication date: 8/12/2015
Collection year: 2015
open-access license: This is an open access article licensed under the terms of the (https://creativecommons.org/licenses/by/4.0/legalcode), which permits unrestricted, noncommercial use, distribution and reproduction in any medium, provided the work is properly cited.
Abstract
Present study efforts are focusing to develop the flurbiprofen fast dissolving tablets using sublimation method to enhance the dissolution rate. In this study an attempt was made to fasten the drug release from the oral tablets by incorporating the sublimating agents in the presence of crosspovidone as superdisintegrant and studied the effect on dissolution rate when compared to conventional tablets. In the present study, sublimated fast dissolving tablets were prepared by direct compression method. The prepared tablets were characterized for physical parameters and drug release behavior and the best formulation was subjected to pharmacokinetic studies. From in vitro drug release studies, the formulation F2 showed fast drug release of about 99.94±0.26% in 30 min, and disintegration time 34.42 ± 0.74 sec. The percent drug release in 15 min (Q15) and initial dissolution rate for formulation F2 was 91.46±1.42%, 6.10%/min. The dissolution efficiency was found to be 53.44 and it is increased by 4.5 fold with F2 sublimated tablets. From the pharmacokinetic evaluation, the conventional tablets producing peak plasma concentration (Cmax) was 9023.68±561.83 ng/ml at 3 h Tmax and F2 sublimated tablets showed Cmax 11126.71±123.56 ng/ml at 2 h Tmax. The area under the curve for the conventional and F2 tablets was 30968.42±541.52 and 42973.66±568.13 ng h/ml. Hence, the development of flurbiprofen fast dissolving tablets by sublimation method is a right way to enhance not only the dissolution rate but also the absorption rate.
INTRODUCTION
Among various active pharmaceutical ingredients, poorly water soluble drugs are related to slower rate of absorption from oral route, so, there is a necessity to enhance the dissolution rate of these drugs to ensure maximum therapeutic utility [1, 2]. Formulation of oral solid dosage forms is convenient for many drugs but they are challenging to formulate if the active substance has poor dissolution rate and low bioavailability. Hence, two areas of pharmaceutical research that focus on improving the oral bioavailability of active agents include enhancing solubility and dissolution rate of poorly water-soluble drugs and enhancing permeability of poorly permeable drugs [3]. To overcome such problems various techniques have been introduced to enhance the dissolution rate and solubility of the drug [4-6]. One of the dissolution enhancement methods is the sublimation technique, which is most widely used and industry feasible method to formulate fast dissolving tablets. Sublimation has been used to produce fast dissolving tablets with high porosity. A porous matrix is formed by compressing the volatile ingredients along with other excipients into tablets, which are finally subjected to a process of sublimation [7, 8]. Some of the recent research examples on sublimation method are meclizine hydrochloride [8], ondansetron [9], lovastatin [10] and clonazepam [11].
The present study is intended to enhance the dissolution rate of flurbiprofen (FLB) by sublimation method with the aid of superdisintegrants. In the present study, sublimation fast dissolving tablets were prepared and studied the effect of different sublimating agents in the presence of crosspovidone on the FLB dissolution rate. FLB is one of the widely used non-steroidal anti-inflammatory agents to treat inflammation. It is a white to off-white crystalline powder and practically insoluble in water [12]. Some of the recent research examples for FLB fast dissolving systems are fast disintegrating tablets [12], fast dissolving tablets [13] and solid dispersions [14].
MATERIALS AND METHODS
Materials
FLB was a gift sample from FDC Limited, Mumbai, India. Crosspovidone was gift samples from Matrix laboratories, Hyderabad, India. All other chemicals used were of analytical grade.
Powder Characterization
Powder mixtures of different formulations were evaluated for angle of repose, bulk density, tapped density and Carr’s compressibility index. The fixed funnel method was employed to measure the angle of repose (θ) and it was calculated using the following formula:
Tan θ = h/r
In which, θ is the angle of repose, h is the height of the cone and r is radius of the cone base. To measure the angle of repose, a funnel was fixed to a stand so that the lower tip of the funnel is 2.5 cm above the surface. A graph paper was placed on a flat surface. The powder blend was allowed to fall freely on the graph paper through the funnel, till the tip of the heap formed just touches the funnel. The radius of the heap was noted and from this angle of repose was determined. Angle of repose less than 30o suggests free flowing properties of the material.
The bulk density of a powder is determined by measuring the volume of a known mass of powder sample that may have been passed through a screen, into a 50 ml graduated cylinder. Tapped densities of powder samples were determined by a tap density apparatus (Intelli, Kshitij Innovations, India). The apparatus was set for 500 tappings for 5 min at a stroke height of 20 mm [15]. The Carr’s compressibility index is determined from the bulk and tapped densities and is calculated using the following formulas:
Carr’s Index = ((ρtap - ρb) / ρtap) / ×100
In which, ρb is bulk density and ρtap is tapped density.
Preparation of Sublimated Fast Dissolving Tablets (FDTs)
FDTs were prepared by sublimation method. Flurbiprofen, sublimating agents, crosspovidone and other tabletting excipients were passed through a mesh no 60. The drug was mixed with proper portion of sublimating agent and superdisintegrant. Care should be taken to confirm the proper mixing with drug. Then excipients other than glidant and lubricant were added and mixed in a poly bag for 5-10 min. The obtained blend was lubricated with talc and magnesium stearate for another 5 min and the resultant mixture was directly compressed into tablets with 8 mm round flat punches using 16 station rotary tabletting machine, Cad mach, Ahmadabad, India. Then these tablets were subjected to sublimation, by placing in a hot air oven at 60°C for 2 h to generate a porous matrix, due to removal of volatilable component. The final weight of the tablet was adjusted to 200 mg and the compositions of the tablets were given in Table 1.
Evaluation of Fast Dissolving Tablets
The prepared tablets were studied for their physical properties like weight variation, hardness and friability. For estimating weight variation, 20 tablets of each formulation were weighed using an electronic weighing balance (AW 120, Shimadzu Corporation, Japan). The strength of tablet is expressed by measuring hardness and friability. The hardness of six tablets was measured using Monsanto tablet hardness tester. Friability was determined on ten tablets in a Roche friabilator (Electrolab, Mumbai, India) for 4 min at 25 rpm.
Determination of Drug Content
For estimation of drug content, ten tablets were crushed in mortar to get powder; this powder was dissolved in 0.1N HCl buffer by sonication for 30 min and filtered through filter paper. The drug content was analyzed spectrophotometrically at 247 nm using UV spectrophotometer. Each measurement was carried out in triplicate and the average drug content was calculated.
In Vitro Disintegration Time and In Vitro Dispersion Time
In vitro disintegration time of FDT’s was determined by following the procedure described by Gohel et al. Briefly, 10 ml of water at room temperature was taken in a petridish of 10 cm in diameter. The tablet was then carefully placed in the centre of petridish and the time required for the tablet to completely disintegrate into fine particles was noted. Measurements were carried out in triplicates [16]. In vitro dispersion time was measured by dropping a tablet in a measuring cylinder containing 6ml of pH 6.8 (simulated saliva fluid). Three tablets from each formulation were randomly selected and in vitro dispersion time is expressed in sec.
Wetting Time
Wetting time was determined as described in the literature elsewhere.Briefly, two circular tissue papers were placed in a Petri dish of 10 cm diameter. Ten ml of water containing 0.5 (% w/v) of phenol red was added to the petridish. The dye solution was used to identify the complete wetting of the tablet surface. A tablet was carefully placed on the surface of the paper in the petridish at room temperature. The time required for water to reach the upper surface of tablet and to completely wet them was noted as wetting time. Wetting time was recorded using stop watch and the measurements were carried out in triplicates.
Water Absorption Ratio (R)
The weight of the tablet prior to placement in the petridish was noted (Wb) using digital balance (Shimadzu, Japan). The wetted tablet was removed and reweighed (Wa). Water absorption ratio (R), was then calculated according to the following equation.
In Vitro Dissolution Study
The release of FLB from FDT’s was carried out using USP XXIV Type II (paddle method) dissolution apparatus (Lab India) at a rotation speed of 100 rpm, and a temperature of 37±0.5°C. The drug release studies were carried out in 0.1N HCl buffer. An aliquot of 5 ml was collected at predetermined time intervals and replaced with fresh dissolution medium. The samples were filtered, by passing through 0.45 µm membrane filters (Millipore, USA) and analyzed spectrophotometrically at 247 nm.
Calculation of Dissolution Parameters
Cumulative percent drug release was plotted as a function of time and percent drug release in 15 min (Q15) was calculated. Initial dissolution rate (IDR) was calculated as percentage dissolved of drug over the first 15 min per min. Dissolution efficiency (DE) was calculated from the area under the dissolution curve at time t and expressed as a percentage of the area of the rectangle described by 100% dissolution in the same time (15). Relative dissolution rate (RDR) is the ratio between amounts of drug dissolved from optimized conventional formulations at 15 min [12].
Drug-Polymer Interaction Studies
To study the possible interaction between FLB and excipients, DSC study was carried out on pure FLB, camphor and best formulation. Differential thermal analysis thermograms were obtained using DSC (Perkin-Elmer, Shelton, U.S). The analyses were performed under nitrogen (nitrogen flow rate 50 ml/min) in order to eliminate oxidative and pyrrolytic effects at a standard heating rate of 15ºC/minute over a temperature range of 50ºC - 350ºC. The FTIR spectra of FLB, camphor and best formulation recorded between 400 to 4000 cm-1 on FTIR to detect the drug-excipients interactions. The FTIR spectra for the test samples were obtained using KBr disk method using an FTIR spectrometer (Perkin Elmer FT-IR, Perkin Elmer Inst. USA). The resultant spectra were compared for any possible changes in the peaks of the spectra.
Stability Studies
To assess the drug and formulation stability, stability studies were done according to ICH guidelines. Best formulation was sealed in aluminum packaging coated inside with polyethylene, and three replicates were kept in the humidity chamber maintained at 40±2oC and 75±5% RH for six months [17]. Samples were collected after six months of storage and analyzed for the drug content and in vitro dissolution rateand they were subjected to statistical analysis using paired t-test to test the significance of difference at 0.05 level of significance (LS). Then the similarity factor (f2) was calculated between dissolution rates of optimized tablets before and after storage to prove the stability of the dosage form [18].
In Vivo Bioavailability Studies
The institutional animal ethical committee (Approval No. SCP/2013/8/2) approved the protocol of the in vivo study of FLB sublimated fast dissolving tablets. Twelve male albino rabbits weighing 2.0±0.3 kg were used for this study. In the present study a crossover study was followed in which twelve male albino rabbits were participated and divided into two equal groups (group I and group II). In the first phase of study group I (n=6) received an immediate release tablet (dose 50 mg) whereas group II (n=6) volunteers received fast dissolving tablet (dose 50 mg). The animals were fasted and had free access to tap water from twelve hours before the experiment. The rabbit’s mouth was opened; tongue was elevated and placed the tablet. Small amount of water was added to surface of the tablet before administering. The mouth was closed for 2 min to avoid chewing or swallowing of the tablet. 2 ml of water was administered after dosing. In the second phase of the study, after 15 days washout period, group I received fast dissolving tablet and group II received immediate release tablet. Blood samples were collected at 0, 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, and 24 hr after dosing from marginal vein.
The collected blood samples were centrifuged at 4000 rpm for 15 min and the serum was separated and transferred to 5 ml micro centrifuge tubes. To the 1 ml of above serum 1 ml of acetonitrile was added and centrifuged for 10 min at 3000 rpm and the supernatant liquid was separated and stored at -40oC until the analysis of sample for unchanged drug. An established HPLC method was used to measure the FLB serum concentration [19]. The quantitative determination of FLB in plasma was performed using HPLC method by injecting the supernatant liquid into the HPLC column (loop volume 20 μl and flow rate 1 ml/min). The analysis was performed at ambient temperature and the run time was set to 8 min and the eluents were monitored at 254 nm using UV detector [15, 19].
Pharmacokinetic Analysis
To elucidate the FLB behavior in plasma, the required pharmacokinetic parameters were calculated using FLB plasma concentration-time data. Pharmacokinetic parameters from plasma data were estimated using Kinetica software (Kinetica 20000 version 3.0, Inna Phase Corporation, 2000). Non compartmental analysis was used. From the plot of time versus plasma concentration, the peak plasma concentration (Cmax) and the time to reach peak plasma levels (Tmax) were obtained. From linear part in the elimination phase of a semi-log plot of concentration versus time, the elimination rate constant (Ke) was calculated. Finally the absorption rate constant (Ka) was calculated from the linear part of residual line using residual method. The area under the curve (AUC) was calculated using the trapezoidal rule [20, 21].
Statistical Analysis
The calculated pharmacokinetic parameters of both immediate release and fast dissolving tablets of FLB were subjected to statistical analysis using analysis of variance (ANOVA) to test the significance of difference. A value of P<0.05 was considered statistically significant.
RESULTS
Powder Characterization
The powder mixtures of different formulations were evaluated for angle of repose, bulk density, tapped density and compressibility index and their values were shown in Table 2. Bulk density and tapped density values ranged from 0.321 to 0.343 and 0.382 to 0.414 respectively. The results of angle of repose and compressibility index (%) ranged from 27.66±3.95 to 31.76±0.70 and 13.21 to 21.31 respectively.
Evaluation of Fast Dissolving Tablets
The physical evaluation parameters of FLB fast dissolving tablets were shown in Table 3 and 4. In weight variation test, the pharmacopoeial limit for the tablets of not more than 7.5% of the average weight. The hardness of the tablets was found to be in the range of 2.8 to 3.4 kg/cm2. Another measure of tablets strength is friability. Conventional compressed tablets that loss less than 1% of their weight are generally considered acceptable. The percentage friability for all formulations was below 1% i.e., 0.28 to 0.36, indicating that the friability is within the prescribed limits. The tablets were found to contain 97.24±1.54% to 99.98±0.34% of the labeled amount indicating uniformity of drug content. The disintegration time of all formulations was found in the range of 34.42±0.74 to 61.32±0.14 sec. The dispersion time and wetting time of formulated tablets was found in the range of 62.76±0.62 to 79.46±0.88 and 30.12±1.14 to 44.28±1.68 sec respectively. Water absorption ratio was 40.28±1.37 to 48.22±1.61. From the results it has been found that FDTs containing camphor showed better results than the others.
In Vitro Dissolution Study
From the preliminary studies to optimize the suitable super disintegrating agent and concentration used in the fast dissolving tablets, different formulations were prepared and evaluated for drug in various proportions Fig. (1). From the dissolution studies, the formulation containing 10 mg of crosspovidone showed fast dissolution rate with rapid disintegration time (data is not presented). Among all the formulations, F2 tablets showed complete drug release within 30 min and rapid dissolution when compared to other formulations i.e., 99.94±0.26% in 30 min. While in the similar conditions, the conventional tablets of same dose showed 56.24±0.92% drug release in 60 min Fig. (2).
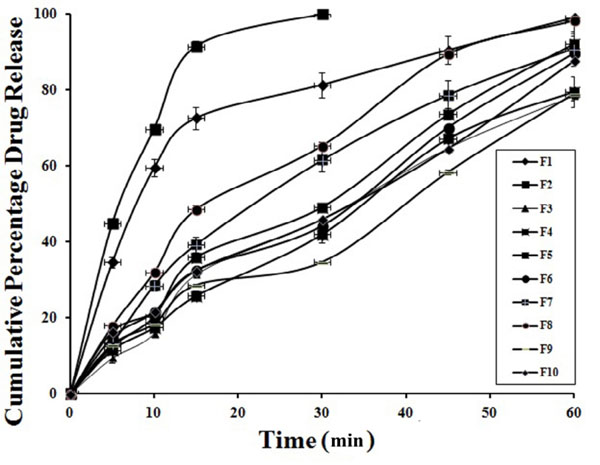 |
Fig. (1). Release profile of FLB from sublimated fast dissolving tablets (n=3). |
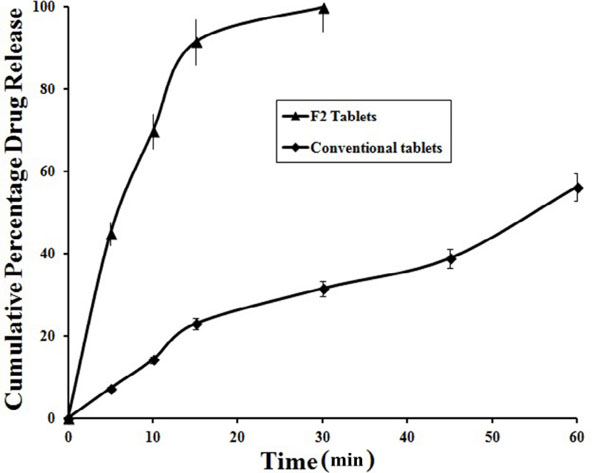 |
Fig. (2). Comparison of FLB release from F2 sublimated fast dissolving tablets and control conventional tablets (n=3). |
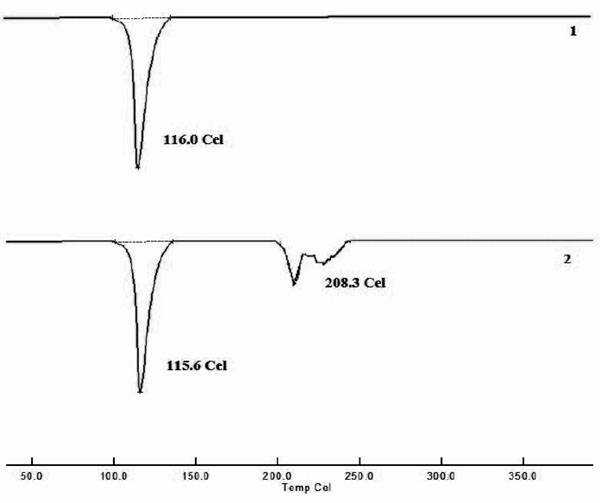 |
Fig. (3). DSC thermograms of 1) FLB, 2) F2 formulation. |
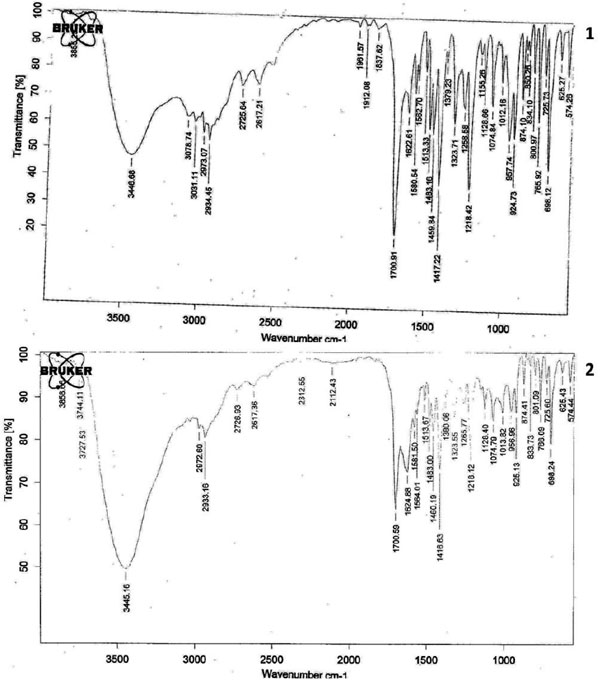 |
Fig. (4). Fourier transform infrared spectra of 1) FLB, 2) F2 formulation. |
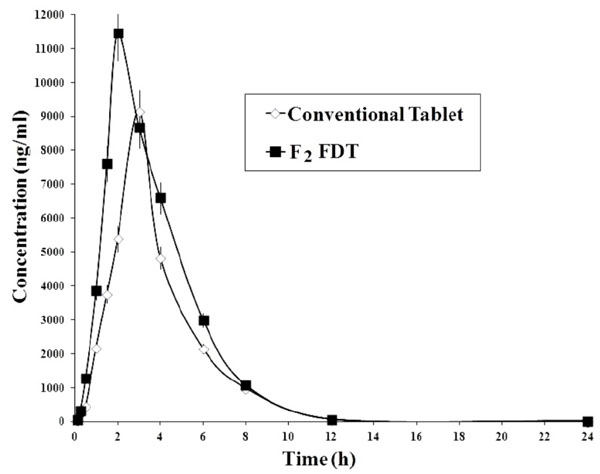 |
Fig. (5). In vivo plasma concentrations of FLB from control conventional and F2 sublimated fast dissolving tablets (n=6). |
Formulation of FLB FDTs using sublimation method.
| Ingredients (mg) | F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 | F10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Flurbiprofen | 50 | 50 | 50 | 50 | 50 | 50 | 50 | 50 | 50 | 50 |
| Camphor | 10 | 20 | - | - | - | - | - | - | - | - |
| Ammonium carbonate | - | - | 10 | 20 | - | - | - | - | - | - |
| Ammonium bicarbonate | - | - | - | - | 10 | 20 | - | - | - | - |
| Benzoic acid | - | - | - | - | - | - | 10 | 20 | - | - |
| Naphthalene | - | - | - | - | - | - | - | - | 10 | 20 |
| Crosspovidone | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| Spray dried lactose | 82 | 72 | 82 | 72 | 82 | 72 | 82 | 72 | 82 | 72 |
| Mannitol | 40 | 40 | 40 | 40 | 40 | 40 | 40 | 40 | 40 | 40 |
| Aspartame | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Aerosil | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| Magnesium stearate | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Total weight | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 |
Characterization of powder mixture.
| Formulation | Angle of Repose*(0) | Bulk Density (g/cc) | Tapped Density (g/cc) | Carr’s Index (%) |
|---|---|---|---|---|
| F1 | 27.92±4.41 | 0.339 | 0.392 | 13.66 |
| F2 | 27.66±3.95 | 0.336 | 0.387 | 13.21 |
| F3 | 31.76±0.70 | 0.331 | 0.401 | 17.35 |
| F4 | 30.56±1.56 | 0.322 | 0.403 | 20.06 |
| F5 | 29.20±3.54 | 0.335 | 0.390 | 14.08 |
| F6 | 29.00±3.12 | 0.321 | 0.408 | 21.31 |
| F7 | 30.05±2.37 | 0.329 | 0.384 | 14.25 |
| F8 | 30.85±0.86 | 0.343 | 0.414 | 17.14 |
| F9 | 29.09±3.91 | 0.324 | 0.382 | 15.14 |
| F10 | 28.76±2.18 | 0.324 | 0.382 | 15.32 |
* All Values Represent Mean ± Standard Deviation, n=3
Physical evaluation of FLB FDTs using sublimation method.
| Formulation | Weight variation*(mg) | Hardness** (kg/cm2 ) |
Friability (%) | Drug content uniformity*** (%) |
|---|---|---|---|---|
| F1 | 204.56±0.64 | 2.9±0.12 | 0.31 | 99.42±0.21 |
| F2 | 199.92±0.73 | 3.1±0.22 | 0.34 | 99.98±0.34 |
| F3 | 201.34±1.06 | 2.8±0.26 | 0.35 | 98.63±1.05 |
| F4 | 199.76±0.46 | 3.4±0.36 | 0.36 | 99.82±0.86 |
| F5 | 202.52±0.24 | 3.2±0.32 | 0.28 | 97.24±1.54 |
| F6 | 204.34±0.52 | 3.1±0.21 | 0.32 | 98.48±0.48 |
| F7 | 206.16±0.64 | 3.4±0.22 | 0.38 | 97.64±1.61 |
| F8 | 198.34±1.12 | 3.3±0.52 | 0.29 | 98.42±0.34 |
| F9 | 199.62±0.48 | 3.1±0.31 | 0.34 | 99.64±0.46 |
| F10 | 202.48±1.42 | 2.9±0.24 | 0.28 | 99.15±1.26 |
* All results correspond to avg ± SD, n=20; ** All results represent avg ± SD, n=6; *** All results represent avg ± SD, n=3
Evaluation of FLB FDTs using sublimation method.
| Formulation | In-vitro Disintegration Time* (sec) |
In-vitro Dispersion Time* (sec) |
Wetting Time* (sec) |
Water Absorption Ratio* |
Q15* |
|---|---|---|---|---|---|
| F1 | 41.36± 0.36 | 69.34±0.56 | 36.42±1.32 | 46.28±1.68 | 72.59±0.21 |
| F2 | 34.42±0.74 | 62.76±0.62 | 30.12±1.24 | 40.28±1.37 | 91.46±1.42 |
| F3 | 46.24±0.24 | 76.31±0.56 | 34.45±1.32 | 42.56±1.52 | 54.82±1.47 |
| F4 | 42.18±0.32 | 78.46±0.24 | 44.28±1.68 | 40.16±1.92 | 62.37±1.87 |
| F5 | 39.28±0.56 | 68.45±0.76 | 30.12±1.14 | 44.26±1.74 | 72.49±0.36 |
| F6 | 59.28±0.52 | 76.48±0.62 | 42.14±1.15 | 48.22±1.61 | 56.34±0.32 |
| F7 | 49.74±0.44 | 74.42±0.45 | 41.38±1.24 | 41.45±1.43 | 72.64±0.56 |
| F8 | 47.42±0.48 | 67.56±0.46 | 34.63±1.46 | 44.28±1.22 | 86.21±1.04 |
| F9 | 61.32±0.14 | 79.46±0.88 | 42.14±1.15 | 44.76±1.44 | 46.88±1.32 |
| F10 | 50.48±0.42 | 76.22±0.43 | 41.38±1.24 | 41.52±1.68 | 49.54±0.23 |
* All results represent avg ± SD, n=3
Dissolution Parameters of FLB F2 fast dissolving and conventional tablets.
| Formulation | (Q15)* | IDR (%/min) | DE | RDR |
|---|---|---|---|---|
| Optimized (F2) | 91.46±1.42 | 6.10 | 53.44 | 3.98 |
| Conventional | 22.92±0.47 | 1.53 | 10.96 |
Stability studies of FLB F2 fast dissolving tablets (n=3).
| Time (min) | Before storage | After 6 months storage |
t-test at 0.05 LS | Similarity Factor (F2) |
|---|---|---|---|---|
| 0 | 0.00±0.00 | 0.00±0.00 | Not Significant | 83.42 |
| 5 | 44.86±0.22 | 42.63±0.36 | ||
| 10 | 69.73±1.14 | 68.12±0.68 | ||
| 15 | 91.46±1.42 | 89.91±0.56 | ||
| 30 | 99.94±0.26 | 97.83±0.43 | ||
| % Assay | 99.98±0.34 | 98.76±0.82 | Not Significant | -- |
Pharmacokinetic parameters of FLB control and F2 fast dissolving tablets (Mean±SD n=6).
| Parameters | Control tablets | F2 fast dissolving tablets | ANOVA test |
|---|---|---|---|
| ka (1/h) | 0.385±0.01 | 0.479±0.01 | Significant |
| ke (1/h) | 0.338±0.01 | 0.362±0.01 | Not Significant |
| Tmax (h) | 3.00±0.01 | 2.00±0.01 | Significant |
| Cmax (ng/ml) | 9023.68±561.83 | 11126.71±123.56 | Significant |
| AUC 0-inf (ng/ml*h) | 30968.42±541.52 | 42973.66±568.13 | Significant |
Calculation of Dissolution Parameters
The percent drug release in 15 min (Q15) and initial dissolution rate for formulation F2 was 91.46±1.42%, 6.10%/min. These were very much higher compared to conventional tablets (22.92±0.47%, 1.53%/min). The improvement in the dissolution characteristics of a drug described in terms of dissolution efficiency (DE) and relative dissolution rate
(RDR). The dissolution efficiency was found to be 53.44 and it is increased by 4.5 fold with F2 FDT tablets compared to conventional tablets i.e., 10.96 and the relative dissolution rate was found to be 3.98 (Table 5).
Drug Polymer Interaction Studies
DSC studies were performed to understand the nature of the drug in the formulated tablets. DSC curves obtained for pure FLB and F2 formulation were shown in Fig. (3). A sharp endothermic peak corresponding to the melting point of Flurbiprofen was found at 116oC whereas during scanning of Camphor and crosspovidone at 217°C, 89.25°C were observed respectively. An endothermic peak corresponding to the melting point of Flurbiprofen in F2 formulation was observed at 115.4oC. Thermogram of the powder mixture did not show any significant shift in the endothermic peak, indicating that there was no physical change in drug in the FDT’s. FTIR studies were performed to understand the nature of the drug in the formulated tablets. The FTIR spectral analysis of pure FLB and formulation F2 were showed the main peaks at comparable wave numbers (Fig. 4). From the pure FLB FTIR spectra gave the main peaks at of 1701.22, 1415.75, 1217.06, 923.9, 765.7and 696.23 cm-1 wave numbers. In case of F2 formulation, the spectra showed peaks at 1701.22, 1419.61, 1217.06, 925.83, 765.7 and 696.23 cm-1. The excipients used in the tablets responsible for some additional peaks which were absent in pure drug spectra.
Stability Studies
To manifest the prospective utility of the formulation, stability studies were carried out at 40±2°C and 75±5% RH for six months. After storage of six months, the formulation
F2 was subjected to a drug assay and in vitro dissolution studies (Table 6) and from the statistical analysis there was no significant difference between before and after storage (P<0.05). The similarity factor value between dissolution profiles of optimized formulation before and after storage was found to be 83.42, which is more than 50 indicates similarity between the dissolution profile before and after storage [18, 19].
Pharmacokinetic Analysis
In the present study, pharmacokinetic evaluation was carried out for both control tablets and F2 sublimated fast dissolving tablets. The mean FLB plasma concentrations following the oral administration of both tablets were showed in Fig. (5) and the mean pharmacokinetic parameters from the in vivo experiments of both tablets were given in Table 7.
DISCUSSION
The scheme of current investigation is to formulate the FLB sublimated fast dissolving tablets to achieve the therapeutic advantage by fasten the dissolution rate. Before going to tablet compression, the results of angle of repose (<30) and compressibility index (<22) indicates fair to passable flow properties of the powder mixture. All the tablet formulations were complied with pharmacopoeial standards, so all the tablets were with worthy physical attributes. In weight variation test, the pharmacopoeial limit for tablets is not more than 7.5% of the average weight. The average percentage deviation of all tablet formulations was discovered to be inside as far as possible and henceforth all formulations passed the uniformity of weight as per the official requirements of Indian Pharmacopoeia, 1996. From the physical characterization, all the tablet formulations were uniform in hardness, friability and drug content uniformity.
From the disintegration test, formulation F2 containing 10% w/w crosspovidone showed the fastest disintegration (34 sec) and less wetting time (30 sec) as compared to other formulations. In the following reported studies, i.e., formulation of piroxicam fast disintegrating tablets [22] and formulation of ondansetron fast dissolving tablet by camphor sublimation [9], similar type of results were showed with crosspovidone. From the dissolution studies, the formulation containing 10 mg of crosspovidone showed fast dissolution rate with rapid disintegration time (data is not presented). From the in vitro dissolution studies, tablets made from camphor showed fast dissolution rate than other sublimating agents. Among all the formulations, F2 tablets showed complete drug release within 30 min and rapid dissolution. The possible reasons and mechanisms for increased dissolution rates are formation of porous structure on the surface of tablet due to sublimation and the presence of superdisintegrants enhance the water permeation (wicking action) in to the tablet leads to fasten the wetting action, disintegration time and finally causes the fast dissolution rate [23].
Overall increase in the dissolution performance of the optimized formulations described in terms of dissolution parameters (IDR, DE, RDR) compared to control tablet could be due to the lesser disintegration time and increased wettability and dispersibility of tablets. The optimized formulation showed 4.5 times improvement in dissolution parameters in comparison to conventional tablets. Similar type of improvement in IDR, DE, RDR was reported in the study of Vemula et al. i.e., flurbiprofen fast disintegrating tablets [12]. From the DSC and FTIR results, it was proved that there was no possibility for drug-excipients interactions. The stability studies data of F2 formulation revealed that there was no significant change in drug content and dissolution rate of tablets before and after storage. The similarity factor value was found as 83.42, which is more than 50 indicates similarity between the dissolution profile before and after storage.
To summarize, F2 formulation containing camphor and crosspovidone was considered as the best formulation that fastens the dissolution rate. In the present study, pharmacokinetic evaluation was carried out for both control tablets and F2 sublimated fast dissolving tablets. In the pharmacokinetic evaluation, the following parameters were calculated. In these, Ka indicates absorption rate and Ke indicates the elimination rate. The Tmax represents rate of absorption and AUC is related to extent of absorption while Cmax is related to both [24].
From these results, rise in the Ka and Ke values was observed in F2 sublimated fast dissolving tablets in contrast to control tablets, which indicates the improvement of absorption rate. The F2 sublimated fast dissolving tablets produced peak plasma concentration Cmax was 11126.71±123.56 ng/ml at 2 h Tmax, but they were found to be 9023.68±561.83 ng/ml at 3 h Tmax, in case of control tablets. This indicates the significant increase in bioavailability. From the estimation of AUC, the F2 sublimated fast dissolving and control tablets were 42973.66±568.13 and 30968.42±541.52 ng-h/ml respectively. Similar type of results was reported in Mettu et al.i.e., Formulation, evaluation and pharmacokinetics of flurbiprofen fast dissolving tablets [13]. From these results there was a significant enhancement of AUC of F2 sublimated tablets when compared with control tablets, which proves the improvement of extent of absorption of FLB. From the above results, it was confirmed that the F2 sublimated fast dissolving tablets showed significant enhancement in rate and extent of absorption of FLB in contrast to control tablets. From the statistical analysis of pharmacokinetic parameters by ANOVA test, there was a significant difference observed in the Ka, Cmax, Tmax and AUC but there was no significant difference in Ke that indicates no difference in the elimination rate constant in both tablets but change was observed in not only in the absorption rate constant but also in peak plasma concentration and time to reach peak plasma concentration. From the results there was a significant difference in the Ka, Cmax, Tmax and AUCbetween control tablets and FDTs, indicating the achievement of rapid as well as improvement in dissolution rate and bioavailability of FLB. Thus the development of the sublimated fast dissolving tablets can be a capable choice to gain the fast dissolution and absorption rate for water-insoluble FLB.
CONCLUSION
In the present study, an attempt was made to develop the fast dissolving tablets of flurbiprofen by sublimation method to enhance dissolution rate. Flurbiprofen fast dissolving tablets were successfully formulated by employing direct compression method and evaluated for different parameters, which were found in the acceptable range. From the dissolution studies of the percent drug release in 15 min (Q15) and initial dissolution rate for formulation F2 was 91.46±1.42%, 6.10%/min and very much higher compared to conventional control tablets. The improvement in the dissolution efficiency was increased by 4.5 fold with F2 sublimated fast dissolving tablets compared to conventional tablets and the relative dissolution rate was found to be 3.98. The results of the pharmacokinetics proved that the there was a significant improvement of bioavailability in case of F2 sublimated fast dissolving tablets when compared to control tablets. In conclusion, development of fast dissolving tablets using sublimation method is able to enhance the dissolution rate of flurbiprofen.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
The authors acknowledge the FDC Ltd, Mumbai, India and Matrix laboratories India for gift samples. The authors also thank to Management of St. John College of Pharmacy and Chaitanya College of Pharmacy Education and Research for providing facilities.