RESEARCH ARTICLE
Recent Advances in Solid-State Analysis of Pharmaceuticals
Syed Nasir Abbas Bukhari*, Ng Shin Hwei , Ibrahim Jantan
Article Information
Identifiers and Pagination:
Year: 2015Volume: 2
First Page: 13
Last Page: 20
Publisher Id: PHARMSCI-2-13
DOI: 10.2174/1874844901502010013
Article History:
Received Date: 5/3/2015Revision Received Date: 11/6/2015
Acceptance Date: 17/6/2015
Electronic publication date: 31/7/2015
Collection year: 2015
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
Abstract
Current analytical techniques for characterizing solid-state pharmaceuticals include powder x-ray diffraction, differential scanning calorimetry, thermogravimetric analysis, infrared spectroscopy, Raman spectroscopy, electron microscopy and nuclear magnetic resonance. Powder x-ray diffraction and differential scanning calorimetry are mainstream techniques but they lack spatial resolution. Scanning electron microscopy and micro-Raman spectroscopy provide good chemical and optical characterization but they are not capable of analysing very small nanoparticles. Transmission electron microscopy and nano-thermal analysis can provide explicit characterization of nanoparticles but they are invasive. Nuclear magnetic resonance offers good spatial resolution but its use is mainly limited by poor sensitivity and high costs. In view of the many challenges posed by existing methods, new and novel techniques are being continually researched and developed to cater to the growing number of solid formulations in the pipeline and in the market. Some of the recent advances attained in the solid-state analysis of pharmaceutical are summarized in this review article.
1. INTRODUCTION
It has been known that pharmaceutical solids can exist in more than one form, be it amorphous or crystalline polymorphs. The different solid forms exhibit different physicochemical properties including melting point, solubility, stability, powder flow, tabletting behaviour and dissolution. These properties, in turn, affect bioavailability of the drug and therapeutic outcome in the consumer. It is therefore crucial to fully characterize a solid drug to ensure that the right form is incorporated into the final pharmaceutical product and maintained throughout its shelf life. Typically, the most thermodynamically stable form of the drug is chosen for development into the final product but more recently, metastable forms with higher solubilities have been used to enhance the dissolution or bioavailability of poorly soluble drugs [1].
Current analytical techniques for characterizing solid-state pharmaceuticals include powder x-ray diffraction, differential scanning calorimetry, thermogravimetric analysis, infrared spectroscopy, Raman spectroscopy, electron microscopy and nuclear magnetic resonance. Powder x-ray diffraction and differential scanning calorimetry are mainstream techniques but they lack spatial resolution. Scanning electron microscopy and micro-Raman spectroscopy provide good chemical and optical characterization but they are not capable of analysing very small nanoparticles. Transmission electron microscopy and nanothermal analysis can provide explicit characterization of nanoparticles but they are invasive. Nuclear magnetic resonance offers good spatial resolution but its use is mainly limited by poor sensitivity and high costs. In view of the many challenges posed by existing methods, new and novel techniques are being continually researched and developed to cater to the growing number of solid formulations in the pipeline and in the market. Some of the recent advances attained in the solid-state analysis of pharmaceutical are presented below.
2. DYNAMIC NUCLEAR POLARIZATION (DNP) ENHANCED NMR
Conventional solid-state NMR techniques utilise cross-polarization magic angle spinning (CP/MAS) pulse sequences to obtain high-resolution spectra from dilute, spin-1/2 nuclei. With CP/MAS, the sample is rotated around an axis inclined at a “magic angle” (( = 54(44’) to the magnetic field so that line broadening is significantly suppressed and resolution is greatly enhanced [2]. This to a large extent, overcomes the overlapping problem of active ingredient by excipients in a pharmaceutical formulation. Although cross-polarization considerably enhances the signals in the NMR spectra, active ingredients still produce relatively low signals compared to excipients due to their small fraction in pharmaceutical formulations. This issue of sensitivity is one of the key reasons behind the recent interest in dynamic nuclear polarization (DNP) techniques.
In DNP NMR, the sample is cooled to < 4K in a strong magnetic field in the presence of a polarizing agent such as trityl-type radicals and tyrosyl radicals. Under these conditions, unpaired electrons become strongly polarized, and the polarization can be transferred to nearby atomic nuclei using microwave irradiation. For solid-state analysis, DNP at high magnetic fields requires relatively powerful and stable microwave sources, yet low enough so that the relative positions of electronic and nuclear spin are frozen.Commercial instruments to accommodate this requirement have recently emerged, such as those combining low-temperature MAS with in situ microwave irradiation [3, 4]. Dynamic nuclear polarization (DNP)-enhanced solid-state NMR spectroscopy has been shown to hold great potential for functional studies of membrane proteins at low temperatures due to its great sensitivity improvement [5, 6].
With the availability of such modern instrumentation, DNP is proving to be quite useful in the signal enhancement of spin-1/2 nuclei such as 13C and 15N [7]. The effect of signal enhancement can be appreciated from a study analysing an amorphous solid dispersion containing 30% diflunisal in polyvinylpyrrolidone (PVP) by 13C CP/MAS with DNP (microwaves on) and without DNP (microwaves off) (Fig. 1) [8].
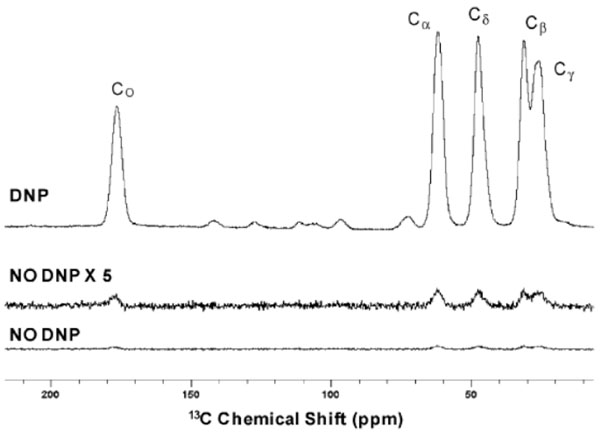 |
Fig. (1). Spectra of 0.5 M U-13C, 15N-proline in glycerol-d8–D2O–H2O (60 : 30 : 10 volume ratio) and 15 mM TOTAPOL biradical, 25 ml sample volume, with DNP (top trace) and without DNP (bottom traces). Reproduced from [8] with permission of The Royal Society of Chemistry. |
 |
Fig. (2). 105 K DNP enhanced natural abundance 13C (left column) and 15N (right column) CPMAS solid-state NMR spectra of (A) crystalline cetirizine dihydrochloride (A), (B) amorphous cetirizine dihydrochloride (A), (C) povidone (P), (D) “LIFE” brand formulation (F1), (E) “CVS” brand formulation (F2), (F) “Reactine” brand formulation (F3), and (G) “Wal-Zyr” brand formulation (F4) impregnated with TCE solutions of TEKPol. "Reprinted (adapted) with permission from [9]. Copyright (2015) American Chemical Society." |
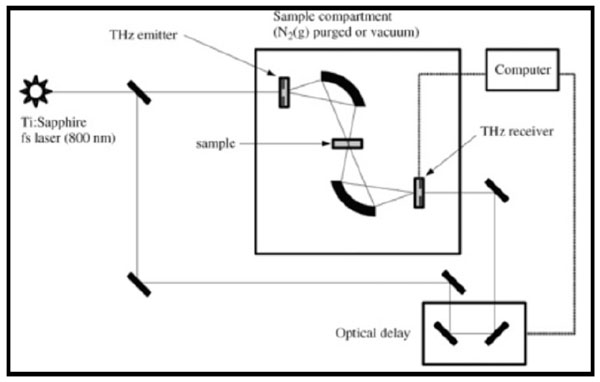 |
Fig. (3). A schematic diagram of a terahertz spectrometer [1]. |
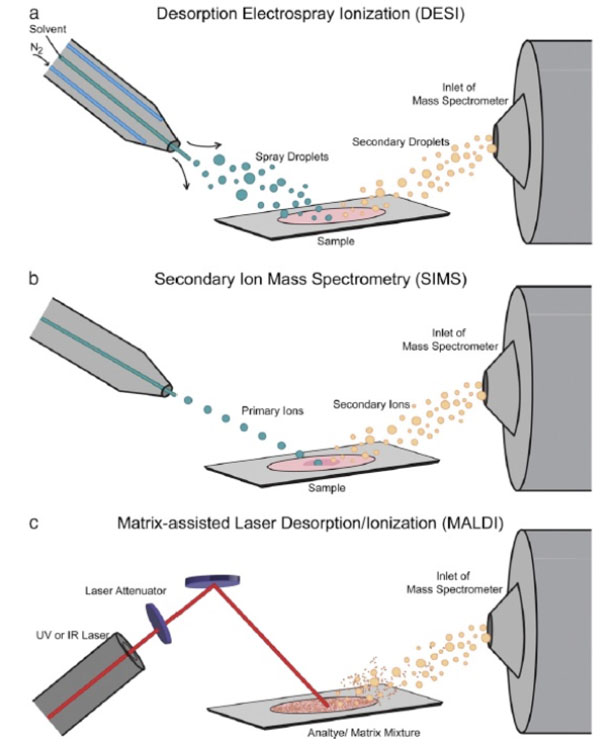 |
Fig. (4). MSI ionization sources. The three most common sources for MSI: (a) Desorption electrospray ionization which uses a stream of solvent ions to desorb and ionize analyte molecules, (b) secondary ion mass spectrometry which uses a beam of ions from an ion gun to sputter analyte ions off the sample, and (c) matrix-assisted laser desorption/ ionization which uses laser irradiation of a matrix-coated sample for desorption and ionization. "Reprinted (adapted) with permission from [21]. Copyright (2015) Elsevier." |
The dispersion was impregnated with bis-TEMPO-bis-ketal radical as polarizing agent and using 1,1,2,2-tetrachloroethane as solvent. A relaxation delay of 7 s was used, during which DNP buildup occurs when microwave irradiation is applied. Spectra were obtained using a MAS rate of 8 kHz with a static field of 9.4 T, at a measurement temperature of 100K. In another more extensive study, the crystalline and amorphous forms of cetirizine, povidone (excipient), as well as four brands of cetirizine tablet formulations (Life, CVS, Reactine and Wal-Zyr) were analysed by both 13C and 15N CP/MAS NMR under DNP (Fig. 2) [9].
The tablets containing 4.8 to 8.7 % w/w of cetirizine dihydrochloride were gently ground by hand with a mortar and pestle and then impregnated with a small volume of 1,1,2,2-tetrachloroethane as solvent and nitroxide biradical TEKPol as polarizing agent. Spectra were obtained using a MAS rate of 8 kHz with a static field of 9.4 T, at a measurement temperature of 105K. It is obvious from the 13C and 15N NMR spectra that all four formulations were of the amorphous form. Sensitivity enhancements of 2 orders of magnitude were obtained with the use of DNP.
3. 19F MAGIC ANGLE SPINNING (MAS) NMR
Due to the properties of 19F which has a relatively high resonance frequency, spin quantum number of ½ and 100% natural abundance [3], 19F NMR is a selective and sensitive multinuclear method for the analysis of complex mixtures of drugs or impurities in pharmaceutical formulations:
Not surprisingly, it is the most common nucleus studied by this technique after 1H and 13C NMR [10]. The instrumentation setup and technique employed in 19F NMR is essentially the same as that of 1H NMR. However, it is better to use a probe specifically designed for fluorine as fluorocarbon polymers often used in the construction of 1H NMR probes can give rise to large, broad, baseline-distorting signals in a 19F NMR spectrum [11]. The advantage of 19F NMR over 1H NMR is that there is no need to suppress the H2O solvent signal hence the use of expensive deuterated solvent is averted. A fluorine nucleus in molecules is on average surrounded by 9 electrons, rather than a single electron as is the case with hydrogen, the range of fluorine chemical shifts and the response of fluorine chemical shifts to the details of the local environment are much higher for fluorine than hydrogen. This means that 19F NMR can be applied not only in the identification and quantification of drug molecules but also in the study of drug-protein binding.
Nowadays, 20-25% of drug in the pharmaceutical pipeline contain at least one fluorine atom12, making 19F NMR an increasingly popular analytical method for pharmaceutical products. A study examining amorphous forms of atorvastatin by 19F MAS NMR showed that acceptable spectra of tablets containing 1-5% active ingredient can be recorded within 10-30 minutes [12]. The study also showed that the results obtained with 19F NMR were closely correlated with that obtained by 13C NMR, FTIR and X-ray diffraction techniques. In another study of amorphous ezetimibe adsorbed onto a mesoporous silica drug delivery system, 19F CP/MAS NMR was found to be a sensitive method for direct detection of the drug
at concentrations of 2 - 16% [13]. Full characterization to determine structural and dynamical properties of the drug as well as interactions between the drug and the silica substrate were successfully carried out. Active ingredients aside, 19F NMR has also been used to quantify polytetrafluoroethylene (PTFE) contamination in drugs that could potentially arise from pharmaceutical manufacturing or testing equipments [14]. The study demonstrated the superb sensitivity of the technique for the detection of fluoropolymer contaminants in drugs at levels as low as 0.02%.
4. NMR MICROIMAGING
In NMR microimaging, the drug is micronized into micro- or nanoparticles then it is embedded in a polymer carrier such as hydroxypropylmellose (HPMC) by means of melting, hot melt extrusion or solvent evaporation before being pressed into a tablet. To observe the hydration and dissolution of the tablet, a small quantity (~ 2 ml) of deuterated water is layered on top of the tablet in the magnet. Then 1H and 2H NMR imaging is performed by fast low angle shot imaging pulse sequence (FLASH) under constant time imaging (CTI). 1H NMR signals are primarily from the solid polymer matrix while 2H NMR signals are due to water. Vertical slices, parallel to the tablet axis, and horizontal slices at different heights within the tablets can be acquired, all with a slice thickness of about 0.5 mm, at different times during the water penetration process. This allows complete profiling of the dissolution or disintegration kinetics of the tablet.
In a study conducted on a 15% antipyrine solid dispersion system using HPMC as the carrier, 1H NMR microimaging was carried out every 4th minute to observe the water distribution in the tablet [15].
In another similar study, 15% flutamide in HPMC solid dispersion tablets were analysed by 1H and 2H NMR for water distribution as well as 19F NMR for selective drug characterization [16] since flutamide possesses 3 fluorine atoms. The study succeeded not only in observing changes in dissolution of the tablet, but also the process of recrystallization and with a very high spatial resolution.
5. SPATIALLY OFFSET RAMAN SPECTROSCOPY (SORS)
Verification of incoming raw materials is a basic regulatory requirement in pharmaceutical manufacturing. To do so, these materials are usually taken through an inspection area and sampled before being analysed by conventional methods such as attenuated total reflectance fourier transform infrared (ATR-FTIR) spectroscopy. This creates a major bottleneck in pharmaceutical manufacturing with associated high costs, the need for highly trained employees and a special chemical handling environment, as well as potential safety hazards to employees. Furthermore, raw materials that are exposed to the air can undergo rapid degradation from moisture, oxygen and light, or could be cross-contaminated by previously inspected materials or sampling tools. In view of these problems, Spatially Offset Raman Spectroscopy (SORS) has recently been proposed as a rapid and non-invasive method to identify raw materials, including API without the need to open the packaging.
In contrast to conventional Raman spectroscopy, spectra of SORS are typically collected from two or more regions that are spatially separated from the laser illumination point. Two Raman spectra are obtained at different spatial offsets, and then processed using a scaled subtraction of one from the other to yield pure Raman spectra of the individual layers. Since light transmits in straight paths through transparent objects but is diffusely scattered through coloured/ opaque/ translucent objects, a special SORS optical arrangement utilizing an obliquely angled laser beam is necessary to cater both the illumination situations. As with conventional Raman spectroscopy and other types of optical spectroscopy, use of SORS is limited to non-metallic containers and containers which are not black or darkly coloured such as cardboard drums. Compared to conventional Raman spectroscopy, however, SORS is capable of overcoming interfering signals and even fluorescence from containers be it transparent or not, thus allowing identification of materials through packaging.
Using SORS, a study was carried out to identify raw materials in a wide variety of packaging materials such as amber glass bottle, thick white plastic high density polyethylene (HDPE) tub, blue plastic sack and multiple layers of coloured paper sacks [17]. Laser beam was delivered to the sample at an angle of ~ 40( then Raman scattered light was collected using a lens, filtered and imaged onto a detector. Zero offset spectra (for container surface) were acquired in < 0.5 s, while offset spectra (for container and content) were obtained between 2 and 10 s. The SORS spectra produced were processed by subtraction of the two spectra to cancel out the container contribution and yield pure spectra of the content. They were then compared with conventional Raman spectra and spectra of the pure material. In all the packaging materials analysed, the processed SORS spectra showed good correlation with the pure material, indicating successful identification and verification of raw materials. This study proves the viability of SORS in the non-invasive and non-destructive identification of pharmaceutical raw materials.
6. SURFACE ENHANCED RAMAN SPECTROSCOPY (SERS) WITH MICROSCOPY
The study on distribution and homogenity of drugs and excipients in solid formulations is an important consideration for a number of reasons. Because distribution of active ingredient in solid dosage forms influences the rate of dissolution, it will affect bioavailability and hence therapeutic outcome. Moreover, distribution of active ingredient in powders determines how homogenous the powder blend will be and eventually influences the uniformity of content of the final product. Detection of counterfeit drugs may also be possible by studying drug distribution as different manufacturing processes give rise to different distributions. Therefore, a combination of techniques comprising surface-enhanced Raman spectroscopy (SERS), microscopic imaging and mapping stage is necessary to obtain spatial images of drug distribution in solid pharmaceutical dosage forms.
SERS is a surface-sensitive technique that enhances Raman scattering by drug molecules adsorbed on rough metal surfaces such as silver. In a study investigating drug distribution in aspirin tablets by SERS, tablets containing aspirin and lactose were prepared by two different processes: “dry technology” and “wet technology” [18]. In the dry technology, both materials were blended in a mortar and pestle while in the wet technology, both materials were blended and dissolved before evaporating the solvent. Tablets were compressed in a KBr disk press and then coated on the top with a silver colloid SERS solution. This will cause aspirin molecules to be adsorbed into the silver particles and enhance the Raman signal. Raman mapping spectra were then collected from 1750-550 cm-1 across a selected area of the tablet and maps of different technologies were compared.
The spatial maps show that the active ingredient (white areas) were homogenously distributed throughout the whole tablet due to wet technology while it was scattered less homogenously in tablets from dry technology. Due to surface enhancement by the silver nanoparticles, trace amount of the drug (0.25%) could be detected, which would not be possible with conventional Raman chemical imaging. SERS also offered the added advantage of a decreased mapping acquisition time as compared with conventional Raman techniques. In the study, mapping for conventional Raman chemical imaging took 14 hours while the SERS chemical imaging only took 20 minutes. Therefore, the study proved the capability of Raman spectroscopy, in particular SERS as a rapid and sensitive tool in drug distribution studies.
7. TERAHERTZ PULSED SPECTROSCOPY COMBINED WITH CHEMOMETRICS
The terahertz region of the electromagnetic spectrum lies between the mid-IR and microwave regions, from 3 - 100 cm-1. Unlike vibrational spectroscopy, molecules excited by terahertz photons undergo torsional low-energy motions. This enables studies on drug polymorphism since the weak intermolecular forces found in crystal lattices are absent in amorphous forms. This means that crystalline forms of a drug will have a well-defined spectrum while the amorphous forms will be essentially invisible in the terahertz region [2]. Apart from that, water shows very strong terahertz signals allowing this technique to be used to determine moisture content in pharmaceuticals [19].
In terahertz pulsed spectroscopy (TPS) Fig. (3) or time-domain terahertz spectroscopy, ultra short laser pulses are generated on the order of femtoseconds. TPS offers the advantages of rapid analysis (full acquisition of spectra in < 1 minute), high sensitivity, low maintenance, suitability for imaging as well as the ability to determine absorption coeffficients and refraction indexes of materials. The relatively low powered laser compared to Raman spectrometers also reduces the risk of thermal-induced changes or photodecomposition of the sample.
Due to the large amount of complex data generated by spectroscopic techniques, chemometrics, a type of multivariate analysis is being increasingly used to analyse them. Qualitative data analysis by principle component analysis (PCA), hierarchical cluser analysis (HCA), linear discriminant analysis (LDA) and soft independent model class
analogy (SIMCA) have proven to be effective techniques for identifying and differentiating between groups of drugs. On the other hand, regression techniques such as multiple linear regression (MLR), principal component regression (PCR) and partial least squares (PLS) regression are vital tools in quantifying and predicting drug content in pharmaceutical formulations. The usefulness of chemometrics lie in its ability to filter out unnecessary information or noise and extract only the essential and relevant portions of the data for analysis.
In a recent study on alkaloids of natural products in solid crystals, terahertz spectroscopy in combination with multiple linear regression were employed for qualitative and quantitative analysis [20]. Different mixtures of the alkaloids were investigated to determine the best combination that would give the lowest signal to noise ratio.
Limit of detection (LOD) was found to be<2 % while limit of quantitation (LOQ) was<5%, indicating the high sensitivity of the method for the analysis of alkaloids in natural pharmaceutical products.
8. MATRIX ASSISTED LASER DESORPTION/ IONIZATION (MALDI) MASS SPECTROMETRY IMAGING (MSI)
In the last decade or so, mass spectrometry imaging (MSI) has garnered considerable interest in the pharmaceutical community [21]. It detects the actual molecules in the image based on their characteristic mass-to-charge ratios (m/z) and does not require the use of labelled compounds such as those used in chemical imaging. In MSI, spectra are collected from the sample, creating a grid of points on the surface of the sample Fig. (4). Then each point is converted into a two-dimensional spatial coordinate before an image is finally constructed by displaying the intensity of a specific m/z at each coordinate. The main advantage of MSI has been mass accuracy, mass resolution and spatial resolution. Mass accuracy describes the agreement of an ion's detected mass to its theoretical mass while mass resolution refers to the minimum difference between two m/z that can be identified as unique ions. Spatial resolution is the minimum distance between two objects in an image at which they can be distinctly discerned.
One of the ionization source technique often employed in pharmaceutical analysis is matrix assisted laser desorption/ ionization (MALDI). In MALDI MSI. The drug is coated with a thin layer of matrix and irradiated with a laser beam. The matrix absorbs much of the energy from the incident laser and provides a very “soft” ionization for the analyte compounds. To produce a good spectra, the matrix must absorb light at the laser wavelength and must not react with the analyte [22].MALDI MSI is capable of imaging molecules small and large, making it a very versatile technique for pharmaceutical drugs.
A study to examine the distribution of active ingredient and excipients in perindopril tablets was carried out using MALDI MSI with multivariate chemometric analysis [22]. The tablet was first eroded to obtain a flat surface for higher quality and reproducibility of spectra acquisition. Then 2,5-dihydroxybenzoic acid and (-cyano-4-hydroxycinnamic acid were deposited by sublimation method onto the tablet before acquiring mass spectra images. This study demonstrated that the use of multivariate algorithms complemented MALDI MSI techniques for explicit identification and examination of drug distribution in tablets.
9. TIME OF FLIGHT SECONDARY ION MASS SPECTROMETRY (TOF-SIMS) IMAGING
Time of flight secondary ion mass spectrometry (ToF-SIMS) imaging is a highly sensitive surface analytical technique which enables examination of complex surfaces of solid pharmaceutical dosage forms. In ToF-SIMS, primary ion pulses are accelerated and focused onto the surface of a sample held under ultra-high vacuum. The bombarding particle impacts the surface and cause a sputter plume containing atoms, whole molecules and fragments of molecules. A certain portion of these will possess a charge that allows
acceleration into a time-of-flight region. Since each ion has equal kinetic energy, their drift velocity will be governed by the relation of kinetic energy to mass. Thus by measuring the time it takes for the secondary ions to reach the detector, the mass-to-charge ratio can be deduced and used to separate them. A diagram depicting the process of ionization is given below [21].
A significant advantage of ToF-SIMS imaging is the acquisition of full mass spectra. This enables the study of any fragment within the mass spectrum, or to extract a spectrum from any region of interest from an image. In addition to that, depth profiling can offer very sensitive compositional and molecular information as a function of depth as well as provide full 3-dimensional distributions within the analysed volume [22, 23]. Time-of-flight instruments have resolving powers ranging from 10,000 to 100,000 and can routinely achieve less than 5 ppm mass accuracy [21]. Moreover, using tandem mass spectrometry (MS/MS) will increase confidence in drug identification and improve the dynamic range of the analysis.
Several studies have used ToF-SIMS to map the distribution of drugs within solid state drug delivery systems by direct analysis of sample cross-sections. Cross-sections of three different controlled release pellet formulations (paracetamol, theophylline and prednisone) were studied with ToF-SIMS imaging and the distribution of drug, excipients and coating characterised [22]. This methodology has also been successfully used to determine the distribution of different drug forms (hydrated and non-hydrated) within a controlled release pellet.
10. . NANOTHERMAL ANALYSIS (NTA) WITH ATOMIC FORCE MICROSCOPY (AFM)
One of the strategies to improving dissolution and bioavailability of poorly soluble drugs is by preparing them in nanoparticles from dried nanosuspensions. However, experimental techniques that can obtain information about the state of nanometer-sized drug particles are scarce [16]. Scanning electron microscopy and micro-Raman spectroscopy provide good chemical and optical characterization but they are not capable of analysing very small nanoparticles. Characterization by conventional powder X-ray diffraction and differential scanning calorimetry (DSC) is limited due to their lack of sensitivity and spatial resolution. Thus nanothermal analysis (nTA) is an emerging technique which combines the high resolution imaging capabilities of atomic force microscopy (AFM) with the ability to characterize the thermal properties of drugs. Compared to its predecessor, scanning thermal microscopy, it offers significantly enhanced spatial resolution.
In nTA the conventional silicon tip used in AFM is replaced by a microfabricated silicon-based probe with a miniature heater that has an imaging spatial resolution of around 5 nm and a thermal property measurement spatial resolution of up to 20 nm [24]. This imaging probe enables utilization of the most widely applied AFM imaging mode, tapping mode, to allow the analysis of softer samples, such as polymers, without damage from the imaging probe. NTA can be used to map thermal properties during imaging, or to carry out local thermal analysis at defined points on a surface. In such analysis, the probe is heated in a temperature cycle similar to DSC whilst in contact with the sample, providing quantitative information on thermal phase transitions. The use of a microfabricated probes is advantageous because of their ability to measure samples down to femtograms and nanoliters. rapid heating and cooling rates exceeding 100oC/s, and high sensitivity to small temperature changes and heat flows.
The application of nTA with AFM can be seen in a study on a nano-dispersed pharmaceutical system containing carbamazepine and hydroxypropylmethyl cellulose (HPMC) [24]. The surface of a 50% carbamazepine-HPMC formulation could be clearly visualised with the nanothermal probe.
In another study on a solid nano-dispersion of felodipine-polyvinylpyrrolidone (PVP), surface morphology could be visibly appreciated by nTA supported by AFM [25]. The formulation was subjected to 95% relative humidity and examined at 0, 1 and 3 days. These studies showed that onset of crystallization can be detected, which is a critical consideration in the stability of poorly soluble drugs formulated in amorphous forms.
11. . SYNCHROTRON RADIATION X-RAY COMPUTED MICROTOMOGRAPHY (SR-CT)
Oral controlled release dosage forms make up a large fraction of the pharmaceutical market due to the ease of administration and patient compliance. The conventional in vitro dissolution tests for controlled release formulations with HPLC or LC/MS analysis can quantify the extent and rate of the drug release. However, they do not provide any insight into the internal structure of the tablet cores. Eventhough tight dissolution specifications are introduced to monitor the quality of the final products, a number of dosage forms fail after approval, and consequently have to be recalled from the market each year [26]. Therefore, modern in vitro microtomography techniques are being utilised as new and efficient tools that can directly reveal the internal structure and dynamic characteristics of the controlled release tablet core at different stages of the drug release process.
In synchrotron radiation computed microtomography (SR-CT), the sample is illuminated by an extended parallel x-ray beam and projection images of the sample are recorded with a 2D detector, commonly a camera coupled with x-ray scintillator. The sample is carefully rotated relative to the X-ray beam and the process is repeated to produce additional two-dimensional images from various viewpoints. Using a sophisticated Fourier transform algorithm, the 2D images are then combined to generate a complete 3D map of the sample. In very simple terms, SR-CT can be thought of as creating a three-dimensional map of the relative atomic density of the sample under evaluation [27]. Accurate sample structure can be reconstructed, given suitable angular sampling. Gray-scale images also can be manipulated using standard image analysis techniques to produce binary or multiple-coloured images and to obtain dimensional information about the sample.Furthermore, SR-CT is a non-destructive technique that has a high penetration ability and provides a reasonable level of resolution (~5–20 mm) [27].
An example of an application can be seen from a study on felodipine sustained release tablets observed at various time points during dissolution [26, 28]. In another study, tablet swelling behaviour of various in-house polymeric preparations was investigated over time using SR-CT. The reconstructed vertical image of a 10% hydroxypropylmethylcellulose (HPMC) + 90% pregelatinised starch tablet showed crack formation after 60 minutes [29].
Besides observing dynamic changes in tablets, SR-CT has also been applied to examine the structures of fine pharmaceutical granules [30]. In the study, two granules were prepared: a bromhexine HCl granule coated with Kollicoat and a wax-matrix acetaminophen granule. It was shown that SR-CT was capable of visualizing the internal structure as well as coating layer of the granules. Moreover, it successfully visualized the high talc regions of the Kollicoat layer that could not be detected by scanning electron microscopy. All these studies prove the high potency of SR-CT in elucidating internal and external shapes and structures of solid pharmaceutical dosage forms.
CONCLUSION
It is evident that recent advances in the analysis of solid-state pharmaceuticals are made possible with the improvement and adaptation of conventional techniques of NMR, Raman, mass spectrometry, thermal analysis and x-ray computed tomography. The progress of such techniques provides critical knowledge and understanding of complex phenomena such as polymorphism, disorder in the crystalline state, molecular-level interactions in crystalline and amorphous materials, and detection of phase impurities. It also enables accurate and vibrant mapping of solid dosage forms at high resolution and expands the capabilities for quantitative analysis of solid formulations even at low levels. The various strengths and weaknesses of each technique can be exploited to achieve full and reliable characterization of solid drugs. It is hoped that the continual progress and advancement in analytical techniques will contribute to better understanding and ultimately effective quality control of solid pharmaceutical dosage forms.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.