RESEARCH ARTICLE
Computational Studies on Imidazo[1,2-a] Pyridine-3-Carboxamide Analogues as Antimycobacterial Agents: Common Pharmacophore Generation, Atom-based 3D-QSAR, Molecular dynamics Simulation, QikProp, Molecular Docking and Prime MMGBSA Approaches
Suraj N. Mali, Hemchandra K. Chaudhari*
Article Information

Identifiers and Pagination:
Year: 2018Volume: 5
First Page: 12
Last Page: 23
Publisher Id: PHARMSCI-5-12
DOI: 10.2174/1874844901805010012
Article History:
Received Date: 28/06/2018Revision Received Date: 29/8/2018
Acceptance Date: 10/9/2018
Electronic publication date: 28/09/2018
Collection year: 2018
open-access license: This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International Public License (CC-BY 4.0), a copy of which is available at: https://creativecommons.org/licenses/by/4.0/legalcode. This license permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Background:
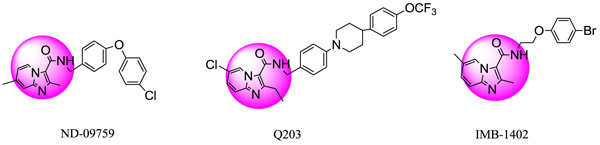
IMB-1402, Q203 and ND09759 analogs were found to have strong efficiency against Multi-drug-resistant tuberculosis (MDR-TB)/Extensively drug-resistant tuberculosis (XDR-TB) strains.
Objectives:
To know the structural necessities for imidazo[1,2-a]pyridine-3-carboxamide analogues, we intended to develop the ligand-based pharmacophore, Quantitative structure–activity relationship models(3D-QSAR model). We also performed Molecular docking, molecular simulation and Prime/Molecular Mechanics Generalized Born Surface Area (Prime/MM-GBSA) studies.
Methods:
All the studies like Common pharmacophore hypothesis generation, Atom based 3D-QSAR study, Prime MMGBSA, Docking, Qikprop, and Molecular dynamics simulation were processed using various modules incorporated within the maestro software interface from Schrodinger, LLC, New York USA (release 2017).
Results:
The common pharmacophore hypothesis(CPH) generation resulted in a five-featured hypothesis HHPRR, containing 1 positive, 2 hydrophobic and 2 aromatic rings. An Atom-based 3D-QSAR model was predicted for twenty seven training sets (a correlation coefficient i.e.R2= 0.9181,Standard deviation i.e.SD =0.3305, variance ratio i.e. F = 85.9) and eleven test sets (cross-validation correlation coefficient i.e.Q2 =0.6745, Root Mean Square Error i.e. RMSE = 0.65, Pearson R = 0.8427, P=1.21E-12) compounds employing alignment based on CPH. The dataset of thirty-eight molecules was allowed for docking into the active site of pantothenate synthetase (PDBID-3IVX) that shows H-bonding (Hydrogen bonding) interactions with residues Gly158, Met195, Pro38 and additionally shows further Pi-cation interactions with a residue like Hie47. We also obtained good simulation results for1.2ns study.
Conclusion:
From the results, the generated 3D-QSAR model may be applicable for additional designing of various novel potent derivatives in the future.