RESEARCH ARTICLE
Structural Insights into the Molecular Design of HER2 Inhibitors
Avinash C. Tripathia, Pankaj Kumar Sonara, Ravindranath Rathoreb, Shailendra K. Sarafa, *
Article Information

Identifiers and Pagination:
Year: 2016Volume: 3
First Page: 164
Last Page: 181
Publisher Id: PHARMSCI-3-164
DOI: 10.2174/1874844901603010164
Article History:
Received Date: 29/12/2014Revision Received Date: 27/06/2016
Acceptance Date: 27/06/2016
Electronic publication date: 29/07/2016
Collection year: 2016
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution-Non-Commercial 4.0 International Public License (CC BY-NC 4.0) (https://creativecommons.org/licenses/by-nc/4.0/legalcode), which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
Abstract
Background:
The present study was aimed at designing some potential candidates as HER2 inhibitors used in breast cancer.
Methods:
An energy optimized pharmacophore (E-pharmacophore) model was developed and used to screen the molecular databases (such as ASINEX and NCI databases) against a six site (ADHRRR) hypothesis. The shape similarity of the retrieved hits was calculated and then filtered applying ADME and Lipinski’s filters. Further, these hits were docked into the crystal structure of HER2 protein (3W32) using Glide XP protocol to obtain the docking poses and XP gscores. The performance of the virtual screening (VS) methods was evaluated using Schrödinger’s decoy set of 1000 molecules. Ranking of the actives in the VS protocol was assessed by a variety of well-established methods including the average rank of actives, EF, ROC, BEDROC, AUAC, and the RIE. The retrieved hits were submitted to Canvas for generating binary fingerprints (dendritic) to identify structural diversity among the hits and clustered on the basis of Tanimoto coefficient using hierarchical clustering.
Results:
Seven structurally diverse clusters were selected applying above protocol, having XP gscores >-10, and fitness scores > 1, considering top scoring cluster representative from each cluster. The best scoring hit 355682-ASINEX was submitted to Combiglide to discover some better candidates with improved scores. Finally, structural interaction fingerprint (SIFT) analysis was employed to study the binding interaction, which showed H-bond interaction with Met793, Gln791 and Thr854 residues of HER2 protein.
Conclusion:
The applied methodology and the retrieved hits could be useful in the design of potent inhibitors of HER2 proteins, commonly found to be expressed in the breast cancer patients.
1. INTRODUCTION
Breast cancer is the second highest occurring cancer in women and one of the leading causes of morbidity and mortality. Breast cancer remains a significant public health problem despite advances in early detection of breast cancer, adjuvant therapy of localized disease, and palliative therapy of metastatic disease. Although, anti-estrogens [1] have provided an effective endocrine therapy, a significant proportion of patients have acquired resistance to these drugs. Hence, the requirement for alternative therapeutics to treat breast cancer has become more urgent. Protein kinases play important roles in signal transduction pathways that regulate numerous cellular functions, including proliferation, differentiation, migration, apoptosis, and angiogenesis. Because signal transduction pathways are up-regulated in many tumor cells, protein kinase inhibitors that target these up-regulated pathways are attractive candidates for cancer therapy [2, 3]. The growth factors and their corresponding receptor tyrosine kinases as well as numerous other protein kinases implicated in malignancies, including non-receptor kinases such as Bcl-Abl and Src kinases, have emerged as important anticancer targets. In addition, the cell cycle regulators (cyclin-dependent kinases, p21 gene) and apoptosis modulators (Bcl-2 oncoprotein, p53 tumor suppressor gene, survivin protein, etc) have also attracted renewed interest as potential targets for anticancer drug discovery. The targeting of human epidermal growth factor receptor (HER) or epidermal growth factor receptor (EGFR) by tyrosine kinase inhibitors (TKIs) represents one such therapeutic approach. The HER kinase family contains four members (EGFR (HER1 or ErbB-1), HER2 (ErbB-2 or neu), HER3 (ErbB-3), and HER4 (ErbB-4)) that are multi-domain proteins consisting of an extracellular ligand binding domain, a single trans-membrane domain, and an intracellular tyrosine kinase domain [4]. Amplification or overexpression of ERBB2 oncogene has been shown to play an important role in the development and progression of certain aggressive types of breast cancer. In recent years, this protein has become an important biomarker and a target of therapy for approximately 30% of breast cancer patients [5]. Several ligands that bind to the extracellular portion of EGFR, HER3, and HER4 have been identified. Upon ligand binding, these receptors form homo- or heterodimers to undergo autophosphorylation of each tyrosine residue within the intracellular kinase domain. Although there are only few known ligands for HER2, this receptor also undergoes spontaneous homo- or heterodimerization and activates downstream signalling [6].
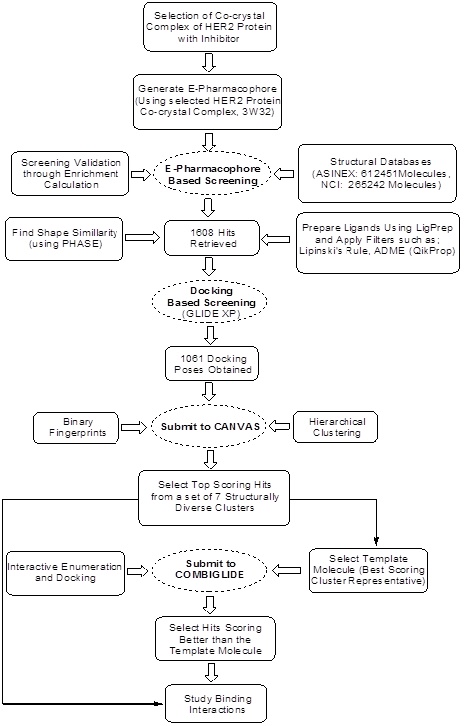 |
Scheme 1. Workflow for the designing of HER2 Inhibitors. |
Pharmacophore based approach has emerged as one of the major tools in drug discovery and is being comprehensively applied in virtual screening (VS), de novo design, lead optimization, multi-target drug design, activity profiling, and target identification and is still in demand for reducing the overall expenditure associated with drug discovery and development. Both, structure and ligand based approaches can be applied parallel to VS, but often these approaches are applied in a stepwise filtering approach [7].
The most commonly applied VS methods are molecular docking, pharmacophore identification and ligand similarity (including shape based), along with a variety of machine learning methods that “learn” to differentiate actives from inactives based on known data [8, 9]. Major challenge in the VS is to create accurate scoring function that can distinguish between novel bioactive and an inactive molecule. Historically, scoring does not correlate well with binding, although consensus scoring, which takes a weighted average of several methods, can result in improvements [10, 11]. In the pharmacophore-based VS approach, a pharmacophore hypothesis is taken as a template to find such molecules (hits) that have chemical features similar to those of the template. Some of these hits might be similar to known active compounds, but some others might be entirely novel in scaffold [12].
The present study was oriented towards the designing of some potential HER2 inhibitors using computations tools. A detailed workflow of the designing has been presented in Scheme 1.
2. MATERIAL AND METHODS
2.1. Compound Dataset
Three crystal structures of pharmaceutically relevant protein target, HER2 in complex with their co-crystal ligands i.e. inhibitors (Scheme 2), having a resolution of less than 2.5Ǻ (PDB-IDs; 3W33 (1.70Å), 3W32 (1.80Å) and 3PP0 (2.25Å)), were considered in the study [13]. Out of these protein-ligand complexes, 3W32 protein-ligand complex was selected on the basis of crystal resolution, re-docking score and RMSD value, for the generation energy optimized structure based pharmacophore model.
 |
Scheme 2. Structure of HER2 co-crystal ligands with their respective PDB-IDs. |
Schrödinger offers the Phase Commercially Available Compound Database (CACDB), containing unique structural records of 612551 molecules. Also, freely available NCI-Open-2012 (265242 molecules as anticancer agents) database was downloaded and used for the VS against an E-pharmacophore model. A widely accepted benchmark dataset of decoys having molecular weight 400Da was downloaded from the Schrödinger’s website [14]. This decoy set consists of 1000 molecules with similar properties but dissimilar topology to the active compounds.
2.2. Protein and Ligand Preparation
Coordinates for each crystal structures were taken from the RCSB Protein Data Bank (PDB) [15] and prepared using the Protein Preparation Wizard, located in Maestro Software Package [16]. Bond orders and formal charges were added for hetero-groups, and hydrogens were added to all atoms in the system. To optimize the hydrogen bond network, His tautomers and ionization states were predicted, 180° rotations of the terminal angle of Asn, Gln, and His residues were assigned, and hydroxyl and thiol hydrogens were sampled. Water molecules in all structures were removed as none of them was found to establish a stable interaction with the protein and the inhibitor. For structures with missing side-chain atoms, the refinement module in Prime [17] was used to predict their conformations. For each structure, a brief relaxation was performed using an all-atom constrained minimization carried out with the Impact Refinement module [18] using the OPLS-2005 force field to alleviate steric clashes that may exist in the original PDB structures. The minimization was terminated when the energy converged or the RMSD reached a maximum cutoff of 0.30 Å.
All the screened hits were prepared by Ligprep module using Epik in Schrödinger package [19] to expand protonation and tautomeric states at 7.0±2.0 pH units, while bioactive conformations of co-crystal ligands were used in the study. Conformational sampling was performed on all database molecules using the ConfGen search algorithm. Confgen with OPLS 2005 force field was applied for generation of conformers. The duplicate poses were eliminated if the RMSD was less than 1.0 Å. A distance dependent dielectric constant of 4 and maximum relative energy difference of 10 kcal/mol was applied as suggested by Salam et.al [20].
2.3. Ligand Docking
Glide energy grids were generated for each of the prepared complexes, and the generated grid file from the prepared receptor was used for docking calculations considering the ligands as flexible but treating the receptor as a rigid structure. The binding site was defined by a rectangular box surrounding the X-ray ligand. Ligands were docked into their respective binding sites in the protein crystal structure. The “Glide XP” protocol [21] was chosen during the docking run which deduces energy terms such as hydrogen-bond interactions, electrostatic interaction, hydrophobic enclosure, and pi–pi stacking interaction and the rest of the parameters were kept at default for the scoring [22].
2.4. Generation of E-pharmacophores
Energy-optimized structure based pharmacophores, i.e. E-pharmacophores were generated through docking, post-processing, E-pharmacophore option situated in scripts menu bar of Maestro Software Package. Glide_XP output (Xpdes (mae.pv)) file, obtained after docking of selected 3W32 HER2 co-crystal ligand to its native protein crystal structure, was used to generate the E-pharmacophore. Pharmacophore sites were automatically generated with Phase [23], using the default set of six chemical features: hydrogen bond acceptor (A), hydrogen bond donor (D), hydrophobic (H), negative ionizable (N), positive ionizable (P), and aromatic ring (R). Each pharmacophore feature site was first assigned an energetic value equal to the sum of the Glide XP contributions of the atoms forming the site. This allows the sites to be quantified and ranked on the basis of these energetic terms involved in their bioactivity towards the target protein.
2.5. Virtual Screening Methods
Three methods of VS protocol were applied in the current study; E-pharmacophore search using Phase [24], docking using Glide [22, 25], and 3D shape similarity search using Phase [26].
2.5.1. E-pharmacophore Based Screening
The E-pharmacophore model of 3W32 HER2 protein-ligand complex was generated and used to screen chemical databases (ASINEX, NCI). For the E-pharmacophore approach, explicit matching was required for the most energetically favorable site, scoring better than -1.0 kcal/mol. Screening molecules were required to match 6 sites for the selected hypotheses Distance matching tolerance was set to 2.0 Å as a balance between stringent and loose-fitting matching alignment. The hit molecules retrieved after E-pharmacophore based search were sorted on the basis of their fitness coefficients.
2.5.2. Shape Screening
After running Phase find matches, a total of 1608 hits were retrieved from ASINEX and NCI databases, which were prepared by LigPrep, and filtered applying Lipinski’s rule and ADME (QikProp) filter criteria. Phase “shape screening” protocol was applied to find out shape similarity of these hits. The active compound 3W32 co-crystal ligand having six pharmacophoric features, along with excluded volume, was selected to create the 3D shape query as the same compound was used to generate the E-pharmacophore in the Phase find matches [26, 27].
2.5.3. Docking Based Screening
Now, these 1608 hits were docked into the crystal structure of 3W32 protein using Glide XP protocol situated in VS Workflows of Maestro user interface of Schrödinger Inc., and the resulting poses were sorted on the basis of their XP gscores.
2.6. Validation of Screening Methods
Many metrics are currently used to evaluate the performance of ranking methods in VS, for instance, the average rank of actives, the enrichment factor (EF), the area under the receiver operating characteristic curve (ROC), Boltzmann-enhanced discrimination of receiver operating characteristic (BEDROC), the area under the accumulation curve (AUAC), the robust initial enhancement (RIE), and were used to determine the robustness of the hypothesis. Evaluating the performance of VS methods is useful to select the method which performs best in a given situation eventually, retrieving active compounds out of a mixed set of active compounds and decoys (compounds presumably inactive against the examined target) [28].
The area under the ROC curve is used by influential groups to measure VS performance in part because it does possess desirable statistical behavior. A value of 1/2 shows that the ranking method does not do better than random picking. It can be interpreted as the probability that an active will be ranked before an inactive. It has a value between 0 (worst performance attainable) and 1 (best performance) [25, 28, 29].
Enrichment factor (EF) is defined as the ratio of the probabilities of searching an active compound in top X% of the data set [30]. A maximum enrichment is therefore 100 if all of the actives (A=1) are found within the top 1% of the decoys (D=0.01).
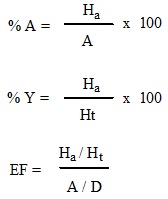 |
Where, Ha: Number of actives in the hit list (true positives); Ht: Number of the hits retrieved; A: Number of active molecules in the database; D: Total number of database compounds; % A: The ratio of the actives retrieved in the hit list (precision); % Y: The yield of actives (recall); EF: Enrichment factor (i.e. enrichment of the concentration of the actives by the model relative to random screening without any pharmacophoric approach); GH: The Guner-Henry score.
 |
Fig. 1. Superposition of the co-crystal ligand 3W32 (Grey) with its docked pose (Sky blue). |
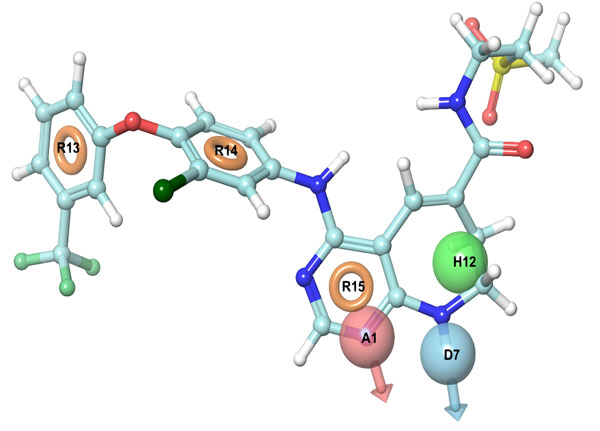 |
Fig. 2a. Energy-optimized pharmacophore (E-pharmacophore) hypotheses ADHRRR (Pink sphere/circle: hydrogen bond acceptor, green sphere/circle: hydrophobic group, orange ring: aromatic ring, light-blue: hydrogen bond donors). |
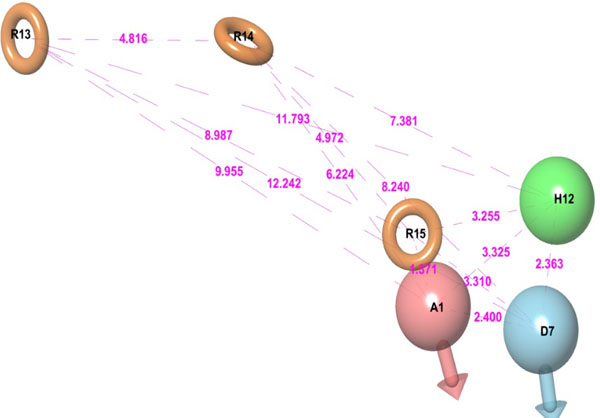 |
Fig. 2b. Site measurement for the selected E-pharmacophore hypothesis ADHRRR (Pink sphere/circle: hydrogen bond acceptor, green sphere/circle: hydrophobic group, orange ring: aromatic ring, light-blue: hydrogen bond donors). |
 |
Figure 3a. ROC Plot between 1-specificity and sensitivity. |
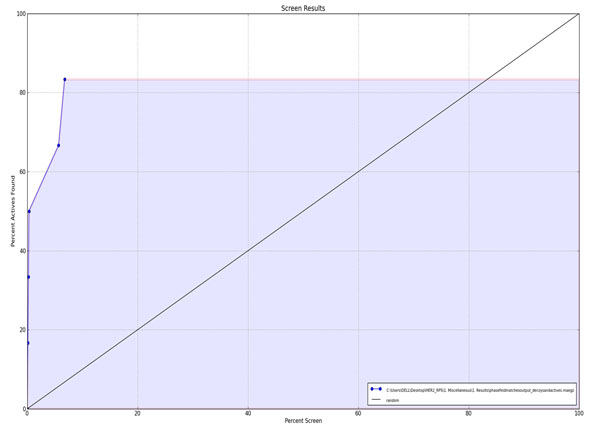 |
Figure 3b. ROC Plot between percent screen and percent actives found. |
 |
Fig. 4a. 2D Interactions of top scoring screened hit 355682-ASINEX at the binding site of HER2 protein, 3W32. |
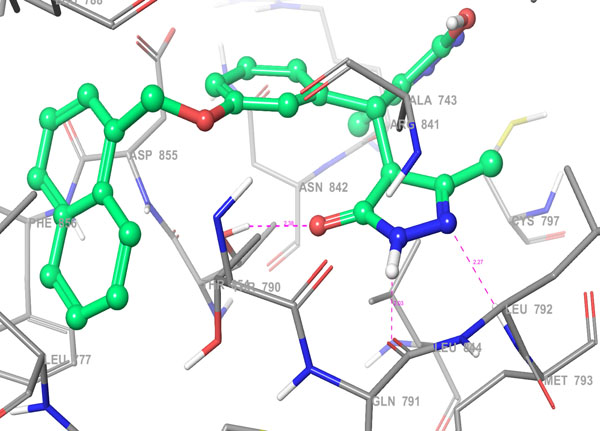 |
Fig. 4b. 3D Interactions (H-bond; Purple colour dotted lines) of top scoring screened hit 355682-ASINEX at the binding site of HER2 protein, 3W32. |
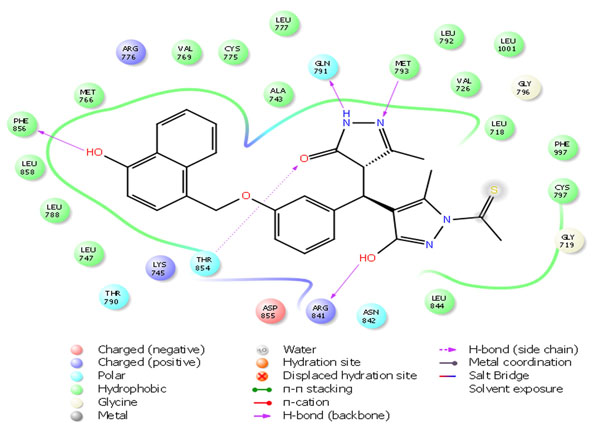 |
Fig. 5a. 2D Interactions of top scoring combinatorial hit 355682-ASINEX_1 at the binding site of HER2 protein 3W32. |
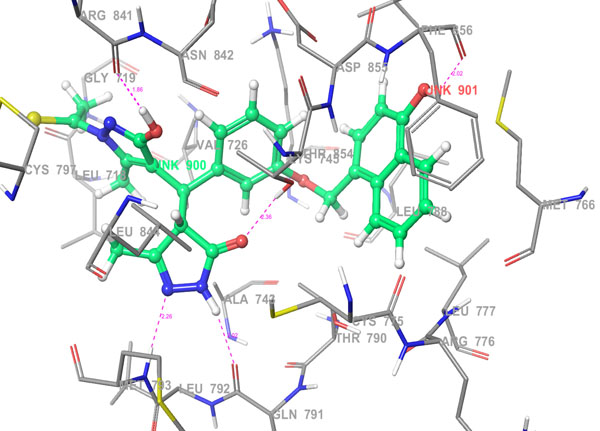 |
Fig. 5b. 3D Interactions (H-bond; Purple colour dotted lines) of top scoring combinatorial hit 355682-ASINEX_1 at the binding site of HER2 protein, 3W32. |
Despite the early recognition problem, the EF has some problems ignoring complete ranking of the whole dataset molecules [30]. A method that is superior to random selection of compounds has EF > 1. To address the problem of EF, as discussed in various reports [31], Sheridan et al. [26] developed an exponential weighted scoring scheme RIE which gives heavier weight in “early recognized” hits. A second enrichment metrics, the BEDROC [28], was also used as a way to ensure that the results and conclusions were significant. BEDROC is a generalization of the ROC that addresses the “early scoring problem” by Boltzmann weighting the hits based on how early they are retrieved. BEDROC can be defined as the probability that an active is ranked before a randomly selected compound was exponentially distributed with parameter α and its values ranges in between 0-1. The value of α= 20.0 is suggested as a reasonable choice for virtual screening evaluations and corresponds to 80% of the total score being accounted for in the top 8% of the database. BEDROC and RIE have a linear relationship.
For the validation of the screening methods, 3 co-crystal ligands, i.e.. inhibitors, present in the crystal structures of HER2 proteins: PDB-IDs; 3W33, 3W32 and 3PP0 (Scheme 2), 23 combinatorial hit molecules (Table 5) and 1000 decoys making a total of 1026 compounds were used in the study. Phase find matches and Glide XP docking was applied selecting these 1026 molecules as input. The output files of these screening methods were chosen to calculate the enrichments using the above said 26 molecules as actives.
2.7. Binary Fingerprints and Hierarchical Clustering Using Canvas
The hit molecules obtained from the above screening methods were submitted to Canvas to generate binary fingerprints (dendritic: linear and branched), using Tanimoto Coefficient (0≤T≤1), to measure the similarity between two fingerprints. The resulted binary fingerprints were used for the clustering of the obtained hits based on their structural similarity. A clustering (hierarchical: non overlapping clustering method where cluster size increases from single molecule per cluster) algorithm divides a group of objects into clusters where molecules within each cluster are more closely related to one another than objects assigned to different clusters. The screened hits (1061) were differentiated into 15 clusters and sorted on the basis of fingerprints and docking XP gscores. The best scoring seven cluster representative molecules were selected, which represent a comprehensive structural diversity amongst the resulted hit molecules.
2.8. In silico ADME Prediction
Nearly 40% of drug candidates fail in clinical trials due to poor ADME (absorption, distribution, metabolism, and excretion) properties. These late-stage failures contribute significantly to the rapidly escalating cost of new drug development. The ability to detect problematic candidates early can dramatically reduce the amount of wasted time and resources, and streamline the overall development process. QikProp [32] program was used for in silico prediction of pharmacokinetic properties of the seven screened hit molecules.
2.9. Generation of Virtual Library Using Combiglide
After binary fingerprinting and hierarchical clustering, the selected best scoring cluster representative was submitted to Combiglide for maximizing the number of hits. A combinatorial library was generated using “Interactive Enumeration and Docking” option present in the Combiglide module of the Schrödinger Software. First, a combinatorial library was created with the help of best scoring template molecule obtained from VS through R group variation, and then it was docked into the crystal structure of the same protein without affecting the previous binding pose. The molecules obtained after combinatorial designing, having better XP gscores than the template molecule, were selected for further studies.
2.10. Structural Interaction Fingerprint (SIFT) Analysis
Docking post-processing, interaction fingerprint option situated in scripts menu bar of Maestro user interface was
used to analyze the structural interactions of the obtained hits (retrieved hits and Combiglide generated combinatorial hits) with the HER2 protein crystal structure.
3. RESULTS AND DISCUSSION
Re-docking (self docking) and cross docking experiments of all the three co-crystal ligands with their respective HER2 proteins and two other protein crystal structures was performed and RMSD (between co-crystal ligand and Glide XP docking pose) was calculated to validate docking efficiency and to find out the best protein crystal structure for further studies. Self docking of 3W32, co-crystal ligand to its native HER2 protein crystal structure (Fig. 1), showed a good XP gscore of -14.874 with appreciable RMSD of 1.4941 (Table 1).
Re-docking/cross docking results of the selected HER2 proteins (PDB-IDs).
| S.No. | Protein (PDB-ID) | Co-Crystal Ligand | Glide XPgscore | RMSD |
|---|---|---|---|---|
| 1. | 3W33 | 3W33 | -13.353 | 1.5096 |
| 2. | “ | 3W32 | -13.753 | 5.6100 |
| 3. | “ | 3PP0 | -13.657 | 5.0509 |
| 4. | 3W32 | 3W32 | -14.874 | 1.4941 |
| 5. | “ | 3W33 | -14.421 | 5.5978 |
| 6. | “ | 3PP0 | -13.885 | 5.5008 |
| 7. | 3PP0 | 3PP0 | -15.263 | 2.3888 |
| 8. | “ | 3W32 | -11.303 | 5.3083 |
| 9. | “ | 3W33 | -12.719 | 5.2553 |
3.1. Virtual Screening
The main purpose of virtual screening is to find a novel scaffold and the possible lead compounds suitable for further development. A combination of structure and ligand based approaches, in association with ligand similarity (shape based) practices, were applied in a stepwise filtering approach to obtain the potential hits.
3.2. E-pharmacophore Based Virtual Screening
It is a hybrid approach of ligand and a structure-based technique which uses docking energy score for finding the bioactive component of ligands against the receptor. Energy optimized six site (one HBdonar, one HBacceptor, one HPhobic, and three ring aromatics) hypothesis (Table 2, Fig. 2) was generated and submitted as a query to retrieve the potential hits from ASINEX and NCI databases. Entire 1608 retrieved hits were subjected to filtering by applying Lipinski’s rule and ADME properties. Database hits were ranked in order of their fitness score (a measure of how well the aligned ligand conformer matches the hypothesis based on RMSD site matching, vector alignments, and volume terms) and selected, having fitness value ≥1. The fitness scoring function is an equally weighted composite of these three terms and ranges from 0 to 3, as implemented in default database screening in Phase [20].
E-pharmacophore hypothesis features (sites) with their respective scores.
| Rank | Feature Label | Score | Score Source |
|---|---|---|---|
| 1 | A1 | -2.17 | PhobEnPairHB + HBond |
| 2 | D7 | -2.08 | PhobEnPairHB + HBond |
| 3 | H12 | -0.74 | PhobeEn + None |
| 4 | R13 | -1.31 | RingChemScoreHphobe |
| 5 | R14 | -1.11 | RingChemScoreHphobe |
| 6 | R15 | -0.84 | RingChemScoreHphobe |
3.3. Validation of Screening Methods
The screening validation of the VS process is needed to check out wether the used method is efficient in retrieving the actives from the databases and ranking them early or not. Different validation parameters were calculated in this study and the results are presented in Table 3. The enrichment results showed that the used screening protocol was satisfactory in retrieving the active compounds from the molecular databases. Three out of the five ranked actives, i.e. in more than 50% yield, were retrieved at 1% of the results with remarkable ROC (0.98), AUAC (0.98), RIE (11.66), GH (0.43) values and a high EF of 50 (at1% of sample size). The ROC plot (Fig. 3a) and % screen plot ROC plot (Fig. 3b) demonstrates that the used method was sensitive and specific in recognizing the active molecules. A substantial value of BEDROC at alpha=20.0, and alpha= 160.9, shows the early recognition of the actives amongst the database compounds.
Virtual screening method validation parameters.
| S. No. | Parameters Used for Screening Validation | Values |
|---|---|---|
| 1. | Number of Actives (1%; 2%; 5%; 10%; 20%) | 3; 3; 3; 5; 5 |
| 2. | % of Actives (1%; 2%; 5%; 10%; 20%) | 50.0; 50.0; 50.0; 83.3; 83.3 |
| 3. | EF (1%; 2%; 5%; 10%; 20%) | 50; 25; 10; 8.3; 4.2 |
| 4. | ROC | 0.98 |
| 5. | RIE | 11.66 |
| 6. | AUAC | 0.98 |
| 7. | BEDROC (alpha=20.0; 160.9) | 0.619; 0.618 |
3.4. Phase Shape Screening
The six structural features (A1, D7, H12, R13, R14, and R15) of 3W32 HER2 protein co-crystal ligand, including excluded volume, were taken as a 3D-shape query. The shape similarity of seven good scoring compounds is presented in the Table 4, which indicate towards the structural diversity amongst the retrieved hits.
3.5. Glide XP Docking
The main aim of docking study is to find the binding affinity between protein–ligand complexes. The hit molecules, retrieved from the virtual screening technique, were further refined by molecular docking using Glide XP protocol to check whether these chemical features mapped with structure-based interaction mode or not. A total of 1061 binding poses were obtained from 1608 screened molecules, which were sorted on the basis of XP gscores (Table 4).
Computational details of seven top scoring screened hit molecules.
| S. No. | Comps. Code | Fitness Coefficient | Matched Ligand Sites | XP gscores | Shape Similarity | SIFT Any Contact |
|---|---|---|---|---|---|---|
| 1. | 355682- ASINEX |
1.349 | A(1) D(7) H(10) R(14) R(12) R(11) | -12.9541 | 0.286 | 2 |
| 2. | 473917- ASINEX |
1.180 | A(1) D(4) H(7) R(12) R(13) R(11) | -12.3129 | 0.292 | 1 |
| 3. | 494978- ASINEX |
1.750 | A(2) D(4) H(7) R(9) R(10) R(11) | -11.7110 | 0.378 | 1 |
| 4. | 461257-ASINEX | 1.100 | A(2) D(3) H(6) R(10) R(8) R(9) | -11.4536 | 0.262 | 1 |
| 5. | 659686-NCI | 1.057 | A(6) D(9) H(10) R(15) R(13) R(12) | -10.6064 | 0.326 | 1 |
| 6. | 632113-NCI | 1.016 | A(3) D(6) H(7) R(13) R(12) R(10) | -10.5978 | 0.296 | 1 |
| 7. | 235809-ASINEX | 1.211 | A(3) D(6) H(9) R(14) R(12) R(10) | -10.2859 | 0.387 | 1 |
3.6. Binary Fingerprints and Hierarchical Clustering
The fingerprints encode the absence or presence of a set of structural fragments (or chemical features) which can be useful in clustering of molecules, diversity analysis and selection of molecules, QSAR model building and similarity search of a database. Out of 15 clusters, 7 structurally diverse representative molecules (Scheme 3) with highest XP gcores (>-10) among the clusters were selected. The structural diversity of the retrieved hits emphasizes the scaffold hoping capability of the E-pharmacophore ADHRRR, when used in the mining of databases of drug like compounds. Also, it has been observed that the entire seven screened hit molecules obtained, satisfy the prerequisites of ADME properties (Table 5).
In silico Predicted pharmacokinetic properties of seven top scoring screened hits using QikProp.
| S. No. | Comps. Code | MW |
donar HB |
accptHB | Volume | PSA |
QlogP o/w Predicted |
metab |
QPlog Khsa |
% Human Oral absorption | Violations of Rule of Five |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1. | 355682- ASINEX |
440.50 | 3 | 5.25 | 1310.31 | 111.49 | 4.011 | 8 | 0.720 | 88.859 | 0 |
| 2. | 473917- ASINEX |
393.53 | 2 | 4.75 | 1293.23 | 73.29 | 4.052 | 9 | 0.853 | 92.347 | 0 |
| 3. | 494978- ASINEX | 398.46 | 1 | 4.5 | 1263.14 | 97.46 | 4.735 | 5 | 0.956 | 100 | 0 |
| 4. | 461257- ASINEX |
352.41 | 1 | 5.5 | 1152.92 | 58.58 | 3.526 | 6 | 0.497 | 92.912 | 0 |
| 5. | 659686-NCI | 560.64 | 2 | 11.5 | 1642.68 | 144.22 | 3.84 | 2 | 0.327 | 73.49 | 1 |
| 6. | 632113-NCI | 474.66 | 2 | 3.5 | 1593.61 | 62.50 | 7.472 | 3 | 1.591 | 100 | 1 |
| 7. | 235809-ASINEX | 502.61 | 1 | 7.5 | 1538.98 | 98.30 | 5.536 | 3 | 1.056 | 82.269 | 2 |
donarHB: Hydrogen bond donar; accptHB: Hydrogen bond acceptor; metab: Number of likely metabolic reactions; QPlogKhsa: Prediction of binding to human serum albumin; Volume: Total solvent accessible volume in cubic angstrom using a probe with 1.4 Ǻ radius; PSA: Van der Waals polar surface area of nitrogen and oxygen atoms; Number of violations of Lipinski’s rule of five.
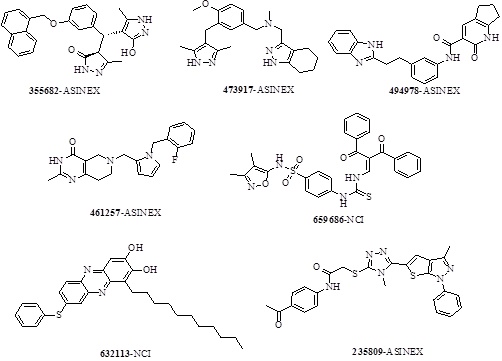 |
Scheme 3. 2D Structure of seven selected top scoring screened hit molecules. |
3.7. Generation of Combinatorial Hits Using Combiglide
The top scoring hit molecule obtained from ASINEX database (355682-ASINEX) was submitted to Combiglide for the interactive enumeration and docking. Variation in R group resulted in the generation of a combinatorial library of more than 10000 combinatorial hits. Out of these, 22 molecules (355682-ASINEX_01 to 355682-ASINEX_22), obtained from varying R1 and R2 substituents, were found to show better binding interactions and XP gscore than the template molecule (355682-ASINEX). The results are summarized in Table 6.
| Comps. Code | Hits obtained from R group variation (Combiglide hits) | Interaction Fingerprints | |||||
|---|---|---|---|---|---|---|---|
| R1 | R2 | Structure | XP gscore | SIFT Any Contact | SIFT Sidechain Interaction |
SIFT HBaccept |
|
| 355682- ASINEX (Template Molecule) |
-H | -H |  |
-12.9541 | 2 | 2 | 0 |
| 355682- ASINEX_1 |
-NH2 | -C6H5 |  |
-14.230 | 3 | 3 | 0 |
| 355682- ASINEX_2 |
-NH2 | -COCH3 | 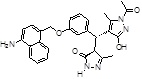 |
-14.108 | 3 | 3 | 0 |
| 355682- ASINEX_3 |
-OH | -CSCH3 | 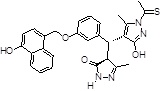 |
-13.948 | 3 | 3 | 0 |
| 355682- ASINEX_4 |
-OH | -COCH3 | 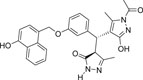 |
-13.926 | 2 | 2 | 0 |
| 355682- ASINEX_5 |
-Cl | -OCH3 | 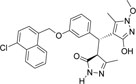 |
-13.913 | 3 | 3 | 0 |
| 355682- ASINEX_6 |
-OCH3 | -CONH2 | 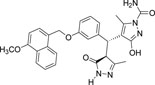 |
-13.790 | 3 | 3 | 0 |
| 355682- ASINEX_7 |
-OH | -OCH3 | 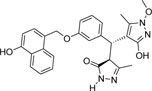 |
-13.720 | 2 | 2 | 0 |
| 355682- ASINEX_8 |
-CONH2 | -C2H5 | 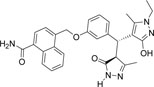 |
-13.706 | 3 | 3 | 1 |
| 355682- ASINEX_9 |
-NH2 | -OCH3 | 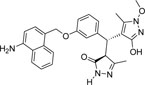 |
-13.705 | 3 | 3 | 1 |
| 355682- ASINEX_10 |
-CH3 | -H | 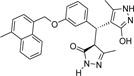 |
-13.698 | 3 | 3 | 0 |
| 355682- ASINEX_11 |
-Cl | -OH | 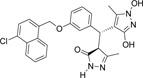 |
-13.615 | 2 | 2 | 0 |
| 355682- ASINEX_12 |
-OCH3 | -CH3 | 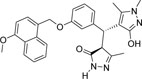 |
-13.588 | 2 | 2 | 0 |
| 355682- ASINEX_13 |
-CH3 | -CSCH3 | 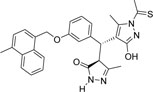 |
-13.558 | 3 | 3 | 0 |
| 355682- ASINEX_14 |
-NH2 | -OH |  |
-13.527 | 2 | 2 | 0 |
| 355682- ASINEX_15 |
-CONH2 | -OCH3 |  |
-13.509 | 2 | 2 | 0 |
| 355682- ASINEX_16 |
-CONH2 | -CSCH3 |  |
-13.432 | 2 | 2 | 0 |
| 355682- ASINEX_17 |
-Cl | -C6H5 |  |
-13.329 | 2 | 2 | 0 |
| 355682- ASINEX_18 |
-C2H5 | -C6H5 |  |
-13.234 | 2 | 2 | 0 |
| 355682- ASINEX_19 |
-C2H5 | -CSCH3 |  |
-13.217 | 2 | 2 | 0 |
| 355682- ASINEX_20 |
-NH2 | -H |  |
-13.115 | 3 | 3 | 0 |
| 355682- ASINEX_21 |
-NH2 | -CONH2 |  |
-13.052 | 3 | 3 | 0 |
| 355682- ASINEX_22 |
-NH2 | -C2H5 | 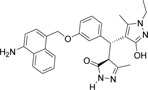 |
-13.019 | 3 | 3 | 0 |
3.8. Binding Interactions and SIFT Analysis
The selected hit molecules were further analyzed by visual inspection and SIFT analysis (Tables 4 and 6) to find out orientation and critical interactions between the receptor and ligand. All the seven screened hit molecules have shown XP gscores of >-10. The best scoring hit molecule, 355682-ASINEX interacts (Fig. 4) with essential amino acid residue Met 793 (backbone), similar to that of the 3W32 co-crystal ligand in the complex with the HER2 protein crystal structure. This molecule was also found to be involved in two additional H-bond interactions with Gln791 (backbone) and Thr854 (side chain) amino acid residues. The best scoring combinatorial hit molecule, 355682-ASINEX_1 generated through Combiglide program, showed the highest XP gscore of -14.230, which was comparable to the binding score of 3W32 HER2 protein co-crystal ligand (-14.874). Furthermore, this molecule showed two new H-bond interactions with Arg841 and Phe856 (Fig. 5) in addition to the above interactions.
CONCLUSION
The present study concludes that the small molecules, which were retrieved by screening of ASINEX and NCI databases against a six site E-Pharmacophore hypothesis ADHRRR, satisfied the necessary conditions such as binding affinity and calculated drug-like properties. The methods used in the screening were validated using different parameters to check their ability to retrieve the active molecules from the databases. The structural diversity has been observed in the compounds and the best scoring hit molecule 355682-ASINEX was virtually populated to some molecules with better binding capability using combinatorial principles. Moreover, these combinatorial hits were showing additional advantage of H-bond interactions along with the side chain interactions over the template molecule. Thus, the obtained hits could be treated as good leads in the design of potent inhibitors of HER2 proteins, most commonly found to be expressed in the breast cancer patients.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
This study was funded by the All India Council for Technical Education (AICTE), New Delhi, India, under Research Promotion Scheme (RPS). The authors are also thankful to the technical support team of Schrodinger Inc. for their valuable suggestions and help during the implementation of the project.
REFERENCES
| [1] | Garay JP, Park BH. Androgen receptor as a targeted therapy for breast cancer. Am J Cancer Res 2012; 2(4): 434-45. |
| [2] | Ishikawa T, Seto M, Banno H, et al. Design and synthesis of novel human epidermal growth factor receptor 2 (HER2)/epidermal growth factor receptor (EGFR) dual inhibitors bearing a pyrrolo[3,2-d]pyrimidine scaffold. J Med Chem 2011; 54(23): 8030-50. |
| [3] | Traxler P. Tyrosine kinases as targets in cancer therapy - successes and failures. Expert Opin Ther Targets 2003; 7(2): 215-34. |
| [4] | Burgess AW, Cho HS, Eigenbrot C, et al. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell 2003; 12(3): 541-2. |
| [5] | Mitri Z, Constantine T, O'Regan R. The HER2 Receptor in breast cancer: Pathophysiology, clinical use, and new advances in therapy. Chemtherapy Res Practice 2012. 743193. |
| [6] | Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. Embo J 2000; 19(13): 3159-67. |
| [7] | Lee HS, Choi J, Kufareva I, et al. Optimization of high throughput virtual screening by combining shape-matching and docking methods. J Chem Inf Model 2008; 48(3): 489-97. |
| [8] | Schierz CA. Virtual screening of bioassay data. J Cheminformatics 2009; 1-21. |
| [9] | Seal A, Passi A, Jaleel UA, Wild DJ. In-silico predictive mutagenicity model generation using supervised learning approaches. J Cheminform 2012; 4(1): 10. |
| [10] | Charifson PS, Corkery JJ, Murcko MA, Walters WP. Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J Med Chem 1999; 42(25): 5100-9. |
| [11] | Liu S, Fu R, Zhou LH, Chen SP. Application of consensus scoring and principal component analysis for virtual screening against beta-secretase (BACE-1). PLoS One 2012; 7(6): e38086. |
| [12] | Yang SY. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov Today 2010; 15(11-12): 444-50. |
| [13] | Kawakita Y, Seto M, Ohashi T, et al. Design and synthesis of novel pyrimido[4,5-b]azepine derivatives as HER2/EGFR dual inhibitors. Bioorg Med Chem 2013; 21(8): 2250-61. |
| [14] | http://www.schrodinger.com/glidedecoyset. |
| [15] | Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res 2000; 28(1): 235-42. |
| [16] | Maestro, v93. New York, NY: Schrodinger, LLC 2012. |
| [17] | Prime, v31. New York, NY: Schrodinger, LLC 2012. |
| [18] | Impact v58. New York, NY: Schrodinger, LLC 2005. |
| [19] | Ligprep v25. New York, NY: Schrodinger, LLC 2012. |
| [20] | Salam NK, Nuti R, Sherman W. Novel method for generating structure-based pharmacophores using energetic analysis. J Chem Inf Model 2009; 49(10): -68 2009; 49(10): 2356-68. |
| [21] | Glide v58. New York, NY: Schrodinger, LLC 2012. |
| [22] | Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. J Med Chem 2004; 47(7): 1739-49. |
| [23] | Phase, v34. New York, NY: Schrodinger, LLC 2012. |
| [24] | Dixon SL, Smondyrev AM, Knoll EH, Rao SN, Shaw DE, Friesner RA. PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J Chem Inf Model 2006; 49(10): 2356-68. |
| [25] | Halgren TA, Murphy RB, Friesner RA, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening J Med Chem 2004; 47(7): 1750-9. |
| [26] | Sheridan RP, Singh SB, Fluder EM, Kearsley SK. Protocols for bridging the peptide to nonpeptide gap in topological similarity searches. J Chem Inf Comput Sci 2001; 41(5): 1395-406. |
| [27] | Tan L, Geppert H, Sisay MT, Gutschow M, Bajorath J. Integrating structure- and ligand-based virtual screening: comparison of individual, parallel, and fused molecular docking and similarity search calculations on multiple targets. ChemMed Chem 2008; 3(10): 1566-71. |
| [28] | Truchon JF, Bayly CI. Evaluating virtual screening methods: good and bad metrics for the "early recognition" problem. J Chem Inf Model 2007; 47(2): 488-508. |
| [29] | Lu SH, Wu JW, Liu HL, et al. The discovery of potential acetylcholinesterase inhibitors: a combination of pharmacophore modeling, virtual screening, and molecular docking studie. J Biomed Sci 2011; 18(8): 488-508. |
| [30] | Zhao W, Hevener KE, White SW, Lee RE, Boyett JM. A statistical framework to evaluate virtual screening. BMC Bioinformatics 2009; 10: 225. |
| [31] | Triballeau N, Acher F, Brabet I, Pin JP, Bertrand HO. Virtual screening workflow development guided by the "receiver operating characteristic" curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4. J Med Chem 2005; 48(7): 2534-47. |
| [32] | QikProp, version 35. New York, NY: Schrodinger, LLC 2012. |